Arine
| Didehydrobenzol als Diradikal (Summenformel: C6H4) |
 ortho-Didehydrobenzol (1,2-Didehydrobenzol) |
 meta-Didehydrobenzol (1,3-Didehydrobenzol) |
 para-Didehydrobenzol (1,4-Didehydrobenzol) |
Arine sind in der Chemie ungeladene, kurzlebige, reaktionsfreudige und cyclische Zwischenprodukte. Sie leiten sich von Aromaten ab, wobei zwei meist orthoständige, benachbarte Substituenten abgespalten wurden und dabei zwei Atomorbitale mit zwei Elektronen resultieren, die auf diese beiden Kohlenstoffatome verteilt sind.[1] Formal weisen ortho-Didehydro-Arine an dieser Stelle eine Kohlenstoff-Kohlenstoff-Dreifachbindung auf. Durch die gespannte Dreifachbindung besitzen solche Arine energetisch sehr tiefliegende LUMOs.[2][3] Daher sind Arine entsprechend reaktiv und neigen zu Additionsreaktionen. In Analogie zu Carbenen und Nitrenen haben Arine einen Singulett- und einen Triplettzustand.
Geschichte
[Bearbeiten | Quelltext bearbeiten]
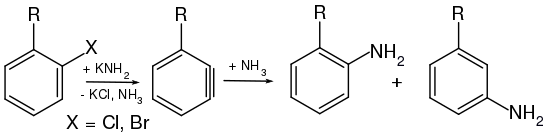
Bereits 1902 postulierten Richard Stoermer und Bruno Kahlert, welche an der Universität in Rostock arbeiteten, dass die Bildung von 2-Ethoxybenzofuran aus der Umsetzung 3-Brombenzofuran mit Basen in Ethanol über die reaktive Zwischenstufe des 2,3-Didehydrobenzofurans abläuft.[4] Auch W. E. Bachmann und H. T. Clarke vermuteten im Jahr 1927 Didehydrobenzol als reaktive Zwischenstufe der Wurtz-Fittig-Synthese.[5] Weitere Untersuchungen erfolgten 1953 durch John Dombrowski Roberts, welcher die Existenz von Arinen durch die Reaktion von 14C-markiertem Chlorbenzol mit Kaliumamid und anschließender Umsetzung mit Ammoniak nachweisen konnte. Dabei entdeckte Roberts, dass die neu eingeführte NH2-Gruppe sich sowohl am 14C-Kohlenstoffatom als auch am benachbarten Kohlenstoffatom befand (cine-Substitution). Die Bildung beider Produkte in gleichen Mengen lässt sich nur durch die Existenz des symmetrischen Intermediats erklären, da die klassische nukleophile aromatische Substitution, welche über einen Additions-Eliminierungsweg verläuft, lediglich das Isomer liefert, in welchem die NH2-Gruppe an das 14C-Atom gebunden ist.[6]

1960 bestätigten Rolf Huisgen und Jürgen Sauer diese Ergebnisse; sie fanden ein Isomerenverhältnis von 48 Prozent zu 52 Prozent, welches sich auf den Isotopie-Effekt zurückführen lässt. Erwin. F. Jenny und John D. Roberts fanden bei der Umsetzung von 14C-markiertem Fluorbenzol mit Phenyllithium ähnliche Ergebnisse. Nach Hydrolyse erhielten sie ein Biphenyl mit einer Umlagerungsrate von 53 Prozent.[7]
Georg Wittig gelang 1956 der Nachweis der reaktiven Arin-Zwischenstufen durch Abfangreaktionen mit Furan in verschiedenen Diels-Alder-Reaktionen.[8] I. P. Fisher und F. P. Lossing fanden in massenspektrometrischen Untersuchungen im Jahr 1963 den Peak von Dehydrobenzol und konnten dessen Ionisationspotential bestimmen.[9] 1969 gelang der erste massenspektroskopische Nachweis von 9,10-Dehydrophenanthren durch Hans-Friedrich Grützmacher und Joachim Lohmann.[10] Der erste infrarotspektroskopische Nachweis von Didehydrobenzol gelang O. L. Chapman durch Photolyse von Phthaloylperoxid, Benzocyclobutadienon oder Phthalsäureanhydrid und Matrix-Isolations-Spektroskopie bei tiefen Temperaturen.[11] Dabei ordnete Chapman der Streckschwingung der C-C-Dreifachbindung fälschlicherweise eine Bande bei 2085 cm−1 zu, welche später von anderen Forschungsgruppen bestätigt wurde. Erst 1992 gelang Juliusz G. Radziszewski der endgültige Nachweis, dass die Streckschwingung der C-C-Dreifachbindung eine Bande bei 1846 cm−1 liefert.[12] Dies entspricht der Erwartung, dass die (formale) C-C-Dreifachbindung durch die Ringspannung schwächer ist als die in ungespannten Alkinen, welche eine Bande um 2150 cm−1 zeigen. Durch Abfangreaktionen mit Furan in einer Diels-Alder-Reaktion konnten Hans F. Ebel und Reinhard W. Hoffmann die Lebensdauer von Didehydrobenzol im Vakuum auf maximal 20 ms berechnen.[13] Ralf Warmuth gelang es schließlich, 1,2-Didehydrobenzol in einem molekularen Container zu isolieren und 1H- und 13C-NMR spektroskopisch zu untersuchen. Durch Vergleich mit Benzol konnten die NMR-Daten wie folgt berechnet werden: 1H-NMR δ(D8THF) = 7,69 ppm und 7,01 ppm; 13C-NMR δ(D8THF) = 182,7 ppm, 126,8 ppm und 138,2 ppm.[14][15]
Die Entdeckung der Arin-Zwischenstufe
[Bearbeiten | Quelltext bearbeiten]Das Postulat einer Arin-Zwischenstufe wurde anhand einer Reihe von Experimenten mit Chlorbenzol (C6H5Cl) aufgestellt. Chlorbenzol, ein recht elektronenreicher Aromat (+M-Effekt des Chlors), erweist sich gegenüber den meisten Nucleophilen wie etwa dem Hydroxidion (OH−) als sehr reaktionsträge; es sind Temperaturen von über 200 °C nötig, um überhaupt eine Reaktion zu generieren. Mit Natriumamid (NaNH2) in flüssigem Ammoniak wird Chlorbenzol bei −33 °C jedoch in Anilin umgewandelt.

Betrachtet man als Ausgangsverbindung nicht Chlorbenzol, sondern p-Chlortoluol (p-CH3C6H4Cl) und setzt sie mit denselben Bedingungen wie beim Chlorbenzol mit Amidionen um, so findet man zwei Produkte: ein erwartetes Amino-Substitutionsprodukt und ein unerwartetes. Somit kann das Chloratom nicht direkt durch die Aminogruppe substituiert worden sein. Zusätzlich findet man bei der Umsetzung von Chlortoluol mit Amidionen immer nur die meta- und die para-Aminoprodukte, niemals aber das ortho-Isomer.

Reaktionsmechanismus
[Bearbeiten | Quelltext bearbeiten]Aus H/D-Austauschreaktionen ist bekannt, dass das Amid-Anion in flüssigem Ammoniak Protonen aus Benzol abstrahieren kann. Besonders rasch geht dieses in o-Stellung zu (−I)-Substituenten, wie z. B. Chlor. Aus diesen Tatsachen lässt sich schließen, dass primär nicht das chlorsubstituierte Kohlenstoffatom des Aromaten angegriffen wird. Das Amidion deprotoniert als starke Base ein alpha-Wasserstoffatom des Chlortoluols, was über ein Carbanion zu einer Dreifachbindungs-Zwischenstufe, dem Arin, führt.

Bei der folgenden Addition von NH3 bildet sich sowohl das m-Isomer, als auch das p-Isomer.

2,6–Dimethylchlorbenzol reagiert unter diesen Bedingungen nicht zum entsprechenden Anilinderivat. 2,6–Dimethylchlorbenzol besitzt keine α-Wasserstoffatome. Somit kann kein Proton abstrahiert werden, das Arin bildet sich nicht aus.
Didehydrobenzol
[Bearbeiten | Quelltext bearbeiten]
Die „zusätzliche“ π-Bindung ist blau markiert.
Das einfachste Arin, C6H4 (mit 1 im rechten Bild bezeichnet), wird manchmal in Anlehnung an die englische Bezeichnung Benzyn genannt. Jedoch muss diese Bezeichnung kritisch betrachtet werden, da es eine spezielle Dreifachbindung impliziert. Ein besserer Name ist Didehydrobenzen beziehungsweise Didehydrobenzol, meist kurz als Dehydrobenzol bezeichnet. Dehydrobenzol ist resonanzstabilisiert, wie die Strukturen 1 und 2 zeigen. Die tatsächliche Verteilung der Elektronen ist aus 3 besser ersichtlich. Die zusätzliche π-Bindung ist in 4a lokalisiert und steht senkrecht zu den π-Bindungen (in 4b) des aromatischen Systems. Dehydrobenzol kann als Diradikal bezeichnet werden: Die „zusätzliche“ π-Bindung in 2, 3, 4a und 4b ist dann homolytisch gespalten, wobei je ein Elektron bei dem Atom steht, das zuvor an der „zusätzlichen“ π-Bindung beteiligt war.
Dehydrobenzol ist aufgrund der Dreifachbindung äußerst reaktiv und hat daher eine sehr kurze Lebensdauer. In Lösungen reagiert es sehr schnell mit vorhandenen Reaktionspartnern und in der Gasphase mit sich selbst zu Di- beziehungsweise Triphenylen.[16] Während in Alkinen (im einfachsten Fall Ethin) normalerweise die unhybridisierten p-Orbitale orthogonal nach oben und hinten zur Molekülachse stehen, was eine optimale Überlappung der Orbitale bewirkt, wird im Arin das p-Orbital verzerrt, um die Dreifachbindung im Ringsystem unterzubringen. Dadurch verringert sich die optimale Überlappung der Orbitale.
Es gibt drei mögliche Diradikale des Dehydrobenzols: 1,2-, 1,3- und 1,4-Didehydrobenzol. Die Bindungsenergien sind in silico 106, 122, und 138 kcal/mol (444, 510, und 577 kJ/mol).[17] Maitland Jones in Princeton hat die möglichen Umlagerungen der 1,2-, 1,3- und 1,4-Didehydrobenzole untersucht.[17][18]
ortho-Didehydrobenzol
[Bearbeiten | Quelltext bearbeiten]Das ortho-Didehydrolbenzol ist das am besten untersuchte Isomer aus der Gruppe der drei möglichen Didehydrobenzole. Auf die Darstellung und seine Reaktionen wird im Folgenden noch entsprechend eingegangen. Wie die hohe Anzahl von Publikationen zeigt, hängt dies sicherlich mit der recht einfachen experimentellen Zugänglichkeit durch eine Vielzahl möglicher Bildungsreaktionen zusammen, aber auch damit, dass sich die beiden Elektronen paaren und dadurch den energetisch günstigeren Singulettzustand erreichen können. Armin Schweig berechnete den Bindungslänge für die C-C-Dreifachbindung im ortho-Didehydrobenzol (je nach Rechenmodell) auf 122 bis 126 pm.[19] Anita M. Orendt ermittelte einen Wert von 124±2 pm. Dies ist ein Wert, der nahe an einer normalen C≡C-Dreifachbindung, wie Ethin (120,3 pm), liegt.[20] Durch η-2 Koordination der C≡C-Dreifachbindung im Metallkomplexen lassen sich ortho-Didehydrobenzole und sogar Tetradehydrobenzol stabilisieren und so gezielt für katalysierte organische Synthesen einsetzen.[21][22][23]
meta-Didehydrobenzol
[Bearbeiten | Quelltext bearbeiten]Im Gegensatz zu den ortho-Didehydroarinen sind die meta- und para-Isomere deutlich instabiler und reaktiver. Entsprechend gelang erst im Jahr 1992 Arbeitsgruppen um Wolfram Sander und Dieter Cremer der erste IR-spektroskopische Nachweis der Existenz von 2,4-Didehydrophenol nach Photolyse von matrixisoliertem Chinondiazid bei 10 K.[24]

Die erste Isolierung von 1,3-Didehydrobezol gelang 1996 in der Arbeitsgruppe um Wolfram Sander durch Photolyse von meta-para-Cyclophan-9,10-dion beziehungsweise durch Gasphasenthermolyse von Diacetylperoxid.[25] Weitere Darstellungswege für meta-Didehydrobenzol sind die Pyrolyse von 1,3-Diiodbenzol oder Blitz-Vakuum-Pyrolyse von 1,3-Dinitrobenzol.[26] Das 1,3-Didehydrobenzol ist unter den gewählten Bedingungen nicht sehr stabil und lagert sich durch eine Ringöffnungsreaktion zum 3-Hexen-1,5-diin um.[4]
para-Didehydrobenzol
[Bearbeiten | Quelltext bearbeiten]

1974 konnten Richard Jones und Robert Bergman zeigen, dass an den Positionen C-1 und C-6 deuteriertes (Z)-Hex-3-en-1,5-diin durch Thermolyse bei 300 °C in ein Gemisch aus 3,4- und 1,6-deuteriertem (Z)-Hex-3-en-1,5-diin übergeht, aber keine 1,3- oder 1,4-deuterierten Isomere gebildet werden. Das Ergebnis lässt sich nur über eine symmetrische und cyclische Zwischenstufe, das 1,4-Didehydrobenzol, erklären.[27]

Die Ringschlussreaktion wird nach seinem Entdecker als Bergman-Cyclisierung bezeichnet. Dass sich das 1,4-Didehydrobenzol tatsächlich bildet, konnte durch entsprechende Abfangreaktionen, beispielsweise durch die Bildung von 1,4-Dichlorbenzol bei Anwesenheit von Tetrachlorkohlenstoff nachgewiesen werden.[27][28] Durch Arbeiten von Kyriacos Costa Nicolaou konnte nachgewiesen werden, dass die Geschwindigkeit der Bergmann-Cyclisierung davon abhängig ist, wie nahe sich die beiden endständigen Kohlenstoffatome (C-1 und C-6) sind.[29] Im (Z)-Hex-3-en-1,5-diin haben die beiden endständigen Kohlenstoffatome einen Abstand von 412 pm, und die Cyclisierung läuft bei 200 °C mit einer Halbwertszeit von 30 s ab. Werden die Enden des En-diins, z. B. durch einen Ringschluss näher zusammen gebracht, so erhöht sich die Reaktionsgeschwindigkeit. Im 3-Cyclodecen-1,5-diin beträgt der Abstand zwischen C-1 und C-6 nur noch 325 pm, und die Bergman-Cyclisierung läuft bereits bei Raumtemperatur mit einer Halbwertszeit von 18 h ab. Aber auch die Gegenwart von Substituenten hat einen Einfluss auf die Geschwindigkeit der Bergmann-Cyclisierung.[30]
Bei der Bergman-Cyclisierung handelt es sich um eine reversible Reaktion, bei der das 1,4-Didehydrobenzol durch eine Retro-Bergmann-Reaktion wieder in das (Z)-Hex-3-en-1,5-diin umgewandelt wird. Da das En-diin gegenüber dem 1,4-Didehydrobenzol zudem das energetisch stabilere Produkt ist, gestaltet sich der direkte Nachweis und die Isolierung des 1,4-Didehydrobenzols entsprechend schwierig. Die Thermolyse von 1,4-Diiodbenzol liefert ausschließlich Hex-3-en-1,5-diin, das 1,4-Didehydrobenzol kann spektroskopisch nicht nachgewiesen werden.[9] Bei einem durch Juliusz G. Radziszewski entwickelten Photolyse-Verfahren konnte 1,4-Didehydrobenzol aber in einer Neonmatrix bei 6 K aus 1,4-Diiodbenzol durch Bestrahlung bei 248 nm dargestellt und charakterisiert werden.[31]
Hetarine
[Bearbeiten | Quelltext bearbeiten]

Als Hetarine werden Arine, in welchen ein Kohlenstoffatom formal durch ein Heteroatom ersetzt wurde, bezeichnet. Auch die Darstellung und Reaktionen der Hetarine wurden in zahlreichen Forschungsarbeiten untersucht.[32][33][34][35][36] Hetarine werden analog den Arinen häufig über Eliminierungsreaktionen halogenierter Heteroaromaten gebildet. Auch in ihren Reaktionen verhalten sich Hetarine ähnlich substituierten Arinen, wobei das Heteroatom einen stärkeren dirigierenden Effekt haben kann. Dabei spielt die Position des Heteroatoms im aromatischen Ringsystem eine entscheidende Rolle. Eine Arbeitsgruppe um Thomas Kauffmann konnte zeigen, dass bei halogenierten Chinolinen Chlor und Brom in der 5-, 6- oder 7-Position nach dem Eliminierungs-Additions-Mechanismus über Arine abgespalten werden, während 8-ständiges Chlor, Brom oder Iod nach dem Additions-Eliminierungsweg ersetzt wird.[37] Bekannte Vertreter der Hetarine sind etwa 3,4-Didehydropyridin, 3,4-Didehydrochinolin oder das oben bereits erwähnte 2,3-Didehydrobenzofuran. Die formale Bildung der Dreifachbindung kann bei Halogenderivaten des Pyridins in 2,3- oder 3,4-Position zum Stickstoffatom erfolgen.
Darstellung
[Bearbeiten | Quelltext bearbeiten]Arine werden größtenteils aus Arylhalogeniden durch Reaktion mit starken Basen gemäß Gleichung (a) dargestellt.[38][39][40] Aber auch andere Reaktionswege wurden beschrieben. So untersuchten Georg Wittig und Reinhard W. Hoffmann die Bildung von Didehydrobenzol durch Zersetzung von Benzothiadiazoloxid bei Raumtemperatur entsprechend Gleichung (b).[41] Martin Stiles und Roy G. Miller berichten über einen ähnlichen Reaktionsweg durch Thermolyse von Benzoldiazonium-o-carboxylat in siedendem Furan nach Gleichung (c).[42][43] Weiter berichteten Georg Wittig und Hans F. Ebel über die Thermolyse von Phthaloylperoxid im Vakuum bei 600 °C gemäß Gleichung (d).[44]

Die Thermolyse von Benzoldiazonioum-o-carboxylat, wie diazotierter Anthranilsäure, nach Gleichung (c) erfolgt dabei über einen konzertierten Mechanismus:

Erzeugung von Dehydrobenzol aus Anthranilsäure.

Auch durch Umsetzung von 1,2-Fluorbrombenzol mit Lithium,[43] Photolysereaktionen oder über Grignard-Verbindungen mit aromatischen Resten[45] sind Arine darstellbar.
Untersuchungen zur Bildung von Arinen aus Arylhalogeniden durch Umsetzung mit Phenyllithium in Diethylether zeigen, dass diese über einen zweistufigen Mechanismus erfolgt.[40][46] Im ersten Schritt wird ein ortho-Wasserstoffatom, welches in unmittelbarer Nachbarschaft zum Halogenatom steht, abgespalten und durch ein Alkalimetall ersetzt. Danach bildet sich im zweiten Schritt das Arin durch Abspaltung des Lithiumhalogenids.

Rolf Huisgen konnte zeigen, dass unter entsprechend gewählten Bedingungen (Phenyllithium in Diethylether) das Fluorbenzol circa 10-mal schneller reagiert als die anderen Arylhalogenide (Fluor >> Brom > Chlor > Iod).[40][46][47] Georg Wittig und Liselotte Pohmer konnten außerdem zeigen, dass die Bildung des Dehydrobenzols umso leichter ist, je elektronegativer das Halogen und je elektropositiver das Metall ist.[48]
Reaktionsgeschwindigkeitskonstanten für die Bildung von Arinen
aus Arylhalogeniden in Diethylether bei 20 °C; 106 · k2 [l · mol−1 · s−1]C6H5F C6H5Cl C6H5Br C6H5I Phenyllithium 40,8 4,0 4,9 2,8 Lithium-piperidid
(ohne Piperidin)860 275 440 168
Werden der Reaktionsmischung Amine zugegeben, zum Beispiel als Lithiumpiperidid/Piperidin, ändern sich die Reaktionsgeschwindigkeiten in Abhängigkeit von der Aminkonzentration. Beim Brombenzol steigt die Reaktionsgeschwindigkeit, während sie umgekehrt bei Fluorbenzol abfällt. Durch eine Verringerung der Aktivierungsenergie der Metallierungsreaktion (1. Schritt) bei Fluoraromaten wird diese im zunehmenden Maße reversibel, wodurch die Bildung des Arins entsprechend verlangsamt, beziehungsweise verhindert wird. Bei der Reaktion von Fluornaphthalin mit Lithiumpiperidid/Piperidin läuft die Substitutionsreaktion entsprechend mit steigendem Piperidingehalt dann zunehmend über den Addition-Eliminierungsmechanismus statt über die Arin-Zwischenstufe ab.[49] Auch die Resistenz von Fluorbenzol gegenüber einer Substitutionsreaktion mit Kaliumamid in Ammoniak kann somit erklärt werden. Durch Untersuchungen an deuteriertem Fluorbenzol konnte gezeigt werden, dass der Metallierungschritt ganz normal ablaufen kann. Das Zwischenprodukt wird danach, durch die hohe Konzentration des Ammoniaks, aber wieder zum (dann entdeuterierten) Fluorbenzol umgewandelt wird.[50] Rolf Huisgen konnte zeigen, dass die Reaktionsgeschwindigkeit der Arinbildung bei Brombenzolderivaten außerdem von weiteren Ringsubstituenten abhängt. So ist die Reaktionsgeschwindigkeit bei einem (weiteren) Bromsubstituenten in ortho-Stellung das 140-Fache und bei einem Fluorsubstituenten das 34-fache, während Alkylreste die Reaktionsgeschwindigkeit reduzieren. Befindet sich der weitere Substituent in meta-Stellung zum Brom, so ist die Reaktionsgeschwindigkeit mit Brom 600-mal, mit Fluor sogar 1700-mal so hoch. Bei einem weiteren Substituenten in para-Stellung zum Brom ist die Änderung der Reaktionsgeschwindigkeit am geringsten.[51]
Reaktionen/Anwendungen
[Bearbeiten | Quelltext bearbeiten]Nukleophile aromatische Reaktionen
[Bearbeiten | Quelltext bearbeiten]Arine können je nach Reaktionspartner entweder als Elektrophile oder als Nukleophile reagieren.[38][52] Als elektronenarme, das heißt elektrophile Zwischenstufen reagieren sie leicht mit Nukleophilen. Rolf Huisgen und Jürgen Sauer konnten zeigen, dass nukleophile, aromatische Reaktionen oft über Arine als Zwischenstufen ablaufen.[40] Der Reaktionsweg über Arine konkurriert dabei mit dem Weg über eine Additions-Eliminierungs-Reaktion. Dabei entscheiden unter anderem die Reaktionsbedingungen und die Ringsubstituenten darüber, welcher der beiden Wege bevorzugt abläuft. So läuft bei der Umsetzung von p-Halogen-Toluolen (Halogen = Chlor, Brom und Iod) mit 4-molarer Natronlauge bei 340 °C zu Kresol praktisch vollständig über eine Eliminierungsreaktion mit einer Arin-Zwischenstufe ab, während bereits bei 250 °C der Additions-Mechanismus einen deutlichen Anteil hat. Ein wichtiges Indiz dafür, welcher Reaktionsmechanismus bei einer aromatischen Substitution abläuft, ist dabei eine Änderung des Substitutionsmusters. Befindet sich die neu eingetretene Gruppe ausschließlich am gleichen Ringatom, kann man von einer klassischen nukleophilen aromatischen Reaktion ausgehen. Bei dieser sind eintretende und austretende Gruppe im Übergangszustand zur gleichen Zeit am gleichen Ringatom gebunden. Bei einer Eliminierungsreaktion über Arine werden stattdessen auch die ortho-ständigen (benachbarten) Ringatome miteinbezogen, wodurch es immer zu einer teilweisen Umlagerung kommt. John D. Roberts untersuchte hierzu Reaktionen an substituierten Arylhalogeniden mit Natrium- oder Kaliumamid in flüssigem Ammoniak zu substituierten Anilinen.[53]
Rest (R) Halogen (X) Ausbeute gesamt [%] Anteil ortho [%] Anteil meta [%] OCH3 Br 33 0 100 CF3 Cl 28 0 100 CH3 Br 64 49 51 CH3 Cl 64 45 55

Kaliumamid kann auch als Katalysator fungieren und die Bildung des Arins beschleunigen. So werden Reaktionen, wie etwa die Umsetzung von Chlorbenzol mit den weniger reaktiven Kaliumanilid, zu Di- oder Triphenylamin überhaupt erst ermöglicht.[54] Albert T. Bottini und John D. Roberts untersuchten die alkalische Hydrolyse von Chlorbenzol [1-14C] und fanden, dass, unter den Bedingungen des technischen Prozesses (15 Prozent NaOH, 370 °C), die Umsetzung bevorzugt über den Eliminierungsprozess mit einem Arin als Zwischenstufe abläuft.[55] Je nach den gewählten Reaktionsbedingungen entstehen in unterschiedlichen Mengen, neben Phenol, auch noch Diphenylether und o- und p-Hydroxybiphenyl.[56]
Ringschlussreaktionen
[Bearbeiten | Quelltext bearbeiten]Rolf Huisgen und H. König berichteten über intramolekulare Ringschlussreaktionen über Arin-Zwischenstufen.[39][57][58] Dabei wurde ein Alkylarylamin zum Beispiel mit Phenyllithium (C6H5Li) umgesetzt. Über das korrespondierende Arin bildet sich in diesem Fall das N-Methyl-2,3-dihydro-indol in 58-prozentiger Ausbeute. Im Gegensatz dazu bildet sich aus der äquivalenten Ausgangsverbindung, in der die Alkylgruppe und das Chloratom in ortho-Stellung zueinander stehen und welches ausschließlich das N-Methyl-2,3-dihydro-indol liefern sollte, eine deutlich geringere Produktmenge.[39]

Enthält der Aminogruppe einen weiteren aromatischen Rest, so können über die Arin-Zwischenstufe Phenanthridine synthetisiert werden.[59]

Ähnlich verlaufen Cyclisierungsreaktionen von o- oder m-Halogen-acylaniliden beziehungsweise -thioacylaniliden mit Kaliumamid (KNH2) unter Bildung eines aromatischen Oxazol- beziehungsweise Thiazol-Ringes.[60]

Diels-Alder Reaktionen
[Bearbeiten | Quelltext bearbeiten]Eine Reaktion mit Arinen als Intermediat ist die Diels-Alder-Reaktion mit Dienen. Innerhalb von wenigen Jahren berichtete Georg Wittig über eine ganze Reihe von Diels-Alder-Reaktion von Arinen, unter anderem mit Cyclopentadien (a),[61][62] Furan (b),[61][8] Pyrrolderivaten (c),[63][61] Benzol (d),[64] Anthracen (e)[61][65] und Cyclohexadien (f).[38] Anthracen wird durch Diels-Alder-Reaktion von Dehydrobenzol mit dem zentralen Benzolring zu Triptycen umgewandelt.[66] Das Pentiptycen ist das Anthracen-Analoge bei der Reaktion mit 1,2,4,5-Tetrabrombenzol und Butyllithium.

Durch Palladium-Katalysatoren können 1,2-Didehydrobenzole mit hoher Regioselektivität zu Triphenylenen trimerisiert werden.[67][68] Bei Anwesenheit von Alkenen oder Alkinen gelingt analog auch die Palladium-katalysierte Synthese von substituierten Phenanthren-Derivaten.[69][70] Mit Vinylindolen reagieren Arine entsprechend unter Bildung von Carbazolen.[71] 1,2,4,5-Tetrabrombenzol reagiert mit Butyllithium und Furan zu 1,4,5,8-Diepoxy-1,4,5,8-tetrahydroanthracen.[72] Die syn- und anti-Stereoisomere können aufgrund der unterschiedlichen Löslichkeiten mit Methanol getrennt werden.

Arylierungsreaktionen
[Bearbeiten | Quelltext bearbeiten]Wird ein Arylhalogenid, wie etwa Chlorbenzol, mit Phenyllithium in siedendem Diethylether umgesetzt, bildet sich daraus Biphenyl, wobei die Reaktionsgeschwindigkeit und die Ausbeute relativ gering ist. Wie weiter oben unter Darstellung aufgeführt, wird das Arin mit Lithiumpiperidid deutlich schneller gebildet. Wird zu dem Gemisch aus Arylhalogenid und Phenyllithium entsprechend Lithiumpiperidid als Katalysator gegeben, wird die Reaktion deutlich beschleunigt und die Ausbeute verbessert.[73]

Durch Variation des Arylchlorids (Aryl1Cl) und der Lithiumaryl-Verbindung (Aryl2Li) eröffnet sich die Möglichkeit, eine ganze Reihe teils ungewöhnlicher Bisaryle zu synthetisieren.[73][74]
Aryl1–Chlorid Aryl2–Lithium Reaktionszeit in h Ausbeute Aryl1–Aryl2 ohne Piperidin mit Piperidin Chlorbenzol Phenyllithium 9 17 61 Chlorbenzol o-Tolyllithium 2 6 23 1-Chlornaphthalin Phenyllithium 2 4 29 1-Chlornaphthalin Phenyllithium 9 22 66 2-Chlornaphthalin Phenyllithium 2 10 50 2-Chlorbiphenyl Phenyllithium 6 18 65 9-Chlorphenanthren Phenyllithium 1 13 58 9-Chlorphenanthren o-Tolyllithium 1 14 60 9-Chlorphenanthren Mesityllithium 40 9 36
Durch Arylierungsreaktionen über Arine lassen sich in guten Ausbeuten sehr sperrige Aryldialkylamine, wie N-Naphthyldiisopropylamin, darstellen, welche auf anderen Wegen nicht zugänglich sind.[75]
Insertionsreaktionen
[Bearbeiten | Quelltext bearbeiten]Mit 2-(Trimethylsilyl)aryltriflaten stehen recht milde Arinvorstufen zur Verfügung, mit denen sich selektive Insertionsreaktionen durchführen lassen. So zeigt die folgende Reaktionsgleichung eine Insertion in eine C-C-Bindung von Malonsäureester.[76]

Eiji Shirakawa zeigte die Möglichkeit zur Insertion von Arinen in die C-N-Bindungen von Harnstoff-Derivaten[77] und Akkattu T. Biju und Frank Glorius berichteten 2010 über hochselektive Insertionsreaktionen von Arinen in die CH-Bindung von Aldehyden. So konnte 4-Brombenzaldehyd mit einer Ausbeute von 98 Prozent zu 4-Brombenzophenon umgesetzt werden.[78] Setzt man 1,2-Didehydrobenzol mit Magnesiumthiolaten um, erhält man die entsprechenden Arylthiolate.[79] Der Magnesiumrest am Ring kann durch entsprechende Elektrophile ersetzt werden.
Dehydrobenzol-Umwandlungen
[Bearbeiten | Quelltext bearbeiten]Die Reaktion von 1,2- zu 1,3-Didehydrobenzol wurde postuliert, um die Pyrolyse (bei 900 °C) der phenylsubstituierten Arinvorstufe 1[17] zu Acenaphthylen 7 zu erklären.

Die Reaktion verläuft über mehrere, reaktive Zwischenprodukte: Das Arin 2 wird aus dem phenylsubstituierten Phthalsäureanhydrid 1 dargestellt und lagert sich unter Ringkontraktion, das heißt Verkleinerung des Cyclohexa- auf den Cyclopenta-Ring, zum Vinyliden 3 um. Das entstandene Carben erfährt eine C-H-Insertionsreaktion zu Pentalen 4 und anschließende Spaltung einer Bindung zu Vinyliden 5. Nach cis-trans-Isomerisation zu 6 folgt eine letzte Insertionsreaktion zur Bildung des Acenaphthylens. Der Beweis der Phenylmigration im Arin 2 aus dem 1,2-Didehydrobenzol zum 1,3-Didehydrobenzol basiert auf Isotopenwanderung. Wird das ipso-Kohlenstoffatom durch ein 13C in der Zwischenstufe ersetzt, wird es bei dem gezeigten Mechanismus im Acenaphthylen an der ipso-Arinposition wieder gefunden. Die Anwesenheit des 13C in der Brückenposition kann nur erklärt werden, wenn 15 Prozent von 2 zu 1,3-Didehydrobenzol A isomerisieren.
Tumortherapie
[Bearbeiten | Quelltext bearbeiten]Die Bildung von para-Didehydrobenzol durch die Bergman-Cyclisierung aus En-Diinen ist von besonderem Forschungsinteresse, da diese den Wirkmechanismus der En-diin-Cytostatika erklären können.[80][81] Die in der Krebsbehandlung eingesetzten Calicheamicine, Esperamicine und Dynemicine wirken über die reaktiven Zwischenstufen von para-Didehydrobenzolderivaten und können gezielt eingesetzt werden, um in vivo die Zuckerketten der DNA-Doppelstränge aufzubrechen, was zum Zelltod führt.[82] Durch deren spezielle Molekülstruktur kann die Bergmann-Cyclisierung bereits bei Raumtemperatur ablaufen.[82] Da die natürlich vorkommenden En-diine stark toxisch sind, müssen Derivate entwickelt werden, die eine maximale Toxizität gegenüber Krebszellen bei geringer Toxizität gegenüber gesunden Zellen haben.[83]
Weitere Möglichkeiten
[Bearbeiten | Quelltext bearbeiten]Die Chemie der Arine wurde angewandt, um neuartige Arylamine in einer Tandem-Reaktion einschließlich zweier Diels-Alder-Reaktionen mit drei Dehydrobenzolmolekülen, die mit einem Imidazol reagieren, zu synthetisieren:[84]

Weblinks
[Bearbeiten | Quelltext bearbeiten]Literatur
[Bearbeiten | Quelltext bearbeiten]- Norman L. Allinger, Michael P. Cava, Don C. de Jongh, C. R. Johnson, N. A. Lebel, Calvin L. Stevens: Organische Chemie. Walter de Gruyter, Berlin 1980, ISBN 3-11-004594-X, S. 650–652.
- Hans Beyer, Wolfgang Walter: Lehrbuch der Organischen Chemie. 19. Auflage. S. Hirzel Verlag, Stuttgart 1981, ISBN 3-7776-0356-2, S. 447–448.
- R. T. Morrison, R. N. Boyd: Lehrbuch der Organischen Chemie. 3. Auflage. Verlag Chemie, Weinheim 1986, ISBN 3-527-26067-6, S. 1131–1136.
- A. Streitwieser. C. H. Heathcock: Organische Chemie. Verlag Chemie, Weinheim 1980, ISBN 3-527-25810-8, S. 1058–1060.
- Kurt P. C. Vollhardt, Neil E. Schore: Organische Chemie. 4. Auflage. Wiley-VCH, Weinheim 2005, ISBN 3-527-31380-X, S. 1170–1172.
- Francis A. Carey, Richard J. Sundberg: Advanced Organic Chemistry, Part A: Structure and Mechanism. 5. Auflage. Plenum, New York, 2007, ISBN 978-0-387-44897-8, S. 821–824.
- Reinhard W. Hoffmann: Dehydrobenzene and Cycloalkynes. Academic Press, 1968, ISBN 0-12-352050-9.
- Paul Rademacher: Vorlesungs-Skript Organische Chemie IV: Reaktive Zwischenstufen (OCIV) (PDF; 4,6 MB)
- Hans Henning Wenk: Matrixisolation Fluorierter Didehydrobenzole und Diradikale. Dissertation. Bochum 2002, DNB 964588234, urn:nbn:de:hbz:294-4854. (PDF; 4 MB)
- Hans Peter Latscha, Uli Kazmaier, Helmut Alfons Klein: Organische Chemie: Chemie-Basiswissen II. 5. Auflage. Springer Verlag, Berlin 2002, ISBN 3-540-42941-7, S. 119–120.
- Peter Sykes: Wie funktionieren organische Reaktionen? 2. Auflage. Wiley-VCH, Weinheim 2001, ISBN 3-527-30305-7.
- Peter Sykes: Reaktionsmechanismen der Organischen Chemie. 7. Auflage. Verlag Chemie, Weinheim 1979, ISBN 3-527-21047-4.
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ T. C. Gilchrist, C. W. Rees: Carbenes, Nitrenes and Arynes. Th. Nelson and Sons, London 1969, ISBN 0-306-50026-4.
- ↑ Stefan Leisering, Christoph A. Schalley: Tutorium Reaktivität und Synthese. Springer-Verlag, Berlin 2017, ISBN 978-3-662-53852-4, S. 188 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Hans Peter Latscha, Uli Kazmaier, Helmut Alfons Klein: Organische Chemie. 6. Auflage. Springer-Verlag, Berlin / Heidelberg 2013, ISBN 978-3-540-77107-4, S. 120, doi:10.1007/978-3-540-77107-4.
- ↑ a b Hans Henning Wenk, Michael Winkler, Wolfram Sander: 100 Jahre Didehydroaromaten. In: Angewandte Chemie. Band 115, Nr. 5, 2003, S. 518–546, doi:10.1002/ange.200390119.
- ↑ W. E. Bachmann, H. T. Clarke: The mechanism of the Wurtz-Fittig reaction. In: J. Am. Chem. Soc. Band 49, 1927, S. 2089–2098, doi:10.1021/ja01407a038.
- ↑ J. D. Roberts, H. E. Simmons, L. A. Carlsmith, C. W. Vaughan: Rearrangement in the reaction of chlorobenzene-1-C14 with potassium amide. In: J. Am. Chem. Soc. Band 75, 1953, S. 3290–3291, doi:10.1021/ja01109a523.
- ↑ Erwin F. Jenny, John D. Roberts: Über den Mechanismus der Bildung von Diphenyl aus Fluorbenzol und Phenyllithium. In: Helvetica Chimica Acta. Band 38, Nr. 5, 1955, S. 1248–1254, doi:10.1002/hlca.19550380520.
- ↑ a b Georg Wittig, Liselotte Pohmer: Über das intermediäre Auftreten von Dehydrobenzol. In: Chemische Berichte. Band 89, Nr. 5, 1956, S. 1334–1351, doi:10.1002/cber.19560890539.
- ↑ a b I. P. Fisher, F. P. Lossing: Ionization Potential of Benzyne. In: J. Am. Chem. Soc. Band 85, 1963, S. 1018–1019, doi:10.1021/ja00890a054.
- ↑ Hans-Friedrich Grützmacher, Joachim Lohmann: Nachweis von 9.10-Dehydro-phenanthren durch Pyrolyse-Massenspektrometrie. In: Justus Liebigs Annalen der Chemie. Band 726, 1969, S. 47–66, doi:10.1002/jlac.19697260109.
- ↑ O. L. Chapman, K. Mattes, C. L. McIntosh, J. Pacansky, G. V. Calder, G. Orr: Photochemical transformations. LII. Benzyne. In: J. Am. Chem. Soc. Band 95, Nr. 18, 1973, S. 6134–6135, doi:10.1021/ja00799a060.
- ↑ Juliusz G. Radziszewski, B. Andes Hess Jr., Rudolf Zahradnik: Infrared spectrum of o-benzyne: experiment and theory. In: J. Am. Chem. Soc. Band 114, Nr. 1, 1992, S. 52–57, doi:10.1021/ja00027a007.
- ↑ Hans F. Ebel, Reinhard W. Hoffmann: Nachweis des Dehydrobenzols in der Gasphase. In: Liebigs Annalen der Chemie. Band 673, Nr. 1, 1964, S. 1–12, doi:10.1002/jlac.19646730102.
- ↑ Ralf Warmuth: 1,2-Didehydrobenzol: ein gespanntes Alkin oder ein Cumulen? — NMR-spektroskopische Charakterisierung in einem molekularen Container. In: Angewandte Chemie. Band 109, Nr. 12, 1997, S. 1406–1409, doi:10.1002/ange.19971091234.
- ↑ Ralf Warmuth: Inner-Phase Stabilization of Reactive Intermediates. In: Eur. J. Org. Chem. Band 2001, Nr. 3, 2001, S. 423–437, doi:10.1002/1099-0690(200102)2001:3<423::AID-EJOC423>3.0.CO;2-2.
- ↑ Frank H. Köhler: Dehydrobenzol: ein (metallorganisches) Stabilisierungsproblem. In: Chemie in unserer Zeit. Band 11, Nr. 6, 1977, S. 190–196, doi:10.1002/ciuz.19770110605.
- ↑ a b c M. E. Blake, K. L. Bartlett, M. Jones Jr.: A m-Benzyne to o-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. In: J. Am. Chem. Soc. Band 125, Nr. 50, 2003, S. 6485–6490, doi:10.1021/ja0213672.
- ↑ A. L. Polishchuk, K. L. Bartlett, L. A. Friedman, M. Jones Jr.: A p-Benzyne to m-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Completion of the Benzyne Cascade. In: J. Phys. Org. Chem. Band 17, 2004, S. 798–806, doi:10.1002/poc.797.
- ↑ Armin Schweig, Norbert Münzel, Hermann Meyer, Andreas Heidenreich: The electronic spectrum of o-benzyne. In: Structural Chemistry. Band 15, Nr. 1, S. 89–100, doi:10.1007/BF00675788.
- ↑ Anita M. Orendt, Julio C. Facelli, Juliusz G. Radziszewski, W. James Horton, David M. Grant, Josef Michl: 13C Dipolar NMR Spectrum of Matrix-Isolated o-Benzyne-1,2-13C2. In: J. Am. Chem. Soc. Band 118, Nr. 4, 1996, S. 846–852, doi:10.1021/ja953417r.
- ↑ Martin A. Bennett, Heinz P. Schwemlein: Metall-Komplexe mit kleinen Cycloalkinen und Arinen. In: Angewandte Chemie. Band 101, Nr. 10, 1989, S. 1349–1373, doi:10.1002/ange.19891011006.
- ↑ F. G. A. Stone: Advances in Organometallic Chemistry, Band 42. Academic Press, 1998, ISBN 0-12-031142-9, S. 148–186 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Sabrina Brait, Stefano Deabate, Selby A. R. Knox, Enrico Sappa: The Coordination and Transformation of Arene Rings by Transition Metal Carbonyl Cluster Complexes. In: Journal of Cluster Science. Band 12, Nr. 1, 2001, S. 139–173, doi:10.1023/A:1016627113620.
- ↑ Götz Bucher, Wolfram Sander, Elfi Kraka, Dieter Cremer: 2,4-Didehydrophenol — erster IR-spektroskopischer Nachweis eines meta-Arins. In: Angewandte Chemie. Band 104, Nr. 9, 1992, S. 1225–1228, doi:10.1002/ange.19921040916.
- ↑ Ralph Marquardt, Wolfram Sander, Elfi Kraka: 1,3-Didehydrobenzol (meta-Benzin). In: Angewandte Chemie. Band 108, Nr. 7, 1996, S. 825–827, doi:10.1002/ange.19961080719.
- ↑ Wolfram Sander, Michael Exner, Michael Winkler, Andreas Balster, Angelica Hjerpe, Elfi Kraka, Dieter Cremer: Vibrational Spectrum of m-Benzyne: A Matrix Isolation and Computational Study. In: J. Am. Chem. Soc. Band 124, Nr. 4, 2002, S. 13072–13079, doi:10.1021/ja012686g.
- ↑ a b Richard R. Jones, Robert G. Bergman: p-Benzyne. Generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-benzenediyl structure. In: J. Am. Chem. Soc. Band 94, Nr. 2, 1972, S. 660–661, doi:10.1021/ja00757a071.
- ↑ Thomas P. Lockhart, Paul B. Comita, Robert G. Bergman: Kinetic evidence for the formation of discrete 1,4-dehydrobenzene intermediates. Trapping by inter- and intramolecular hydrogen atom transfer and observation of high-temperature CIDNP. In: J. Am. Chem. Soc. Band 103, Nr. 14, 1981, S. 4082–4090, doi:10.1021/ja00404a018.
- ↑ K. C. Nicolaou, W.-M. Dai: Chemie und Biologie von Endiin-Cytostatica/Antibiotica. In: Angewandte Chemie. Band 103, Nr. 11, 1991, S. 1453–1481, doi:10.1002/ange.19911031106.
- ↑ Michael Klein: Synthese, Struktur und Eigenschaften funktionalisierter cyclischer Endiine und Untersuchung elektronischer Substituenteneffekte auf die Bergman-Cyclisierung acyclischer Endiine. Dissertation. Regensburg 2003. (PDF; 1,4 MB)
- ↑ Ralph Marquardt, Andreas Balster, Wolfram Sander, Elfi Kraka, Dieter Cremer, J. George Radziszewski: 1,4-Didehydrobenzol. In: Angewandte Chemie. Band 110, Nr. 7, 1998, S. 1001–1005, doi:10.1002/(SICI)1521-3757(19980403)110:7<1001::AID-ANGE1001>3.0.CO;2-D.
- ↑ Th. Kauffmann: Die Hetarine. In: Angewandte Chemie. Band 77, Nr. 13, 1965, S. 557–571, doi:10.1002/ange.19650771302.
- ↑ Thomas Kauffmann, Rolf Wirthwein: Fortschritte auf dem Hetarin-Gebiet. In: Angewandte Chemie. Band 83, Nr. 1, 1971, S. 21–34, doi:10.1002/ange.19710830104.
- ↑ Thomas Kauffmann, Heinz Fischer, Reinhard Nürnberg, Rolf Writhwein: Hetarine, XIV Über die Selektivität hetero- und carbocyclischer Arine gegenüber Basen. In: Justus Liebigs Annalen der Chemie. Band 731, Nr. 1, 1970, S. 23–36, doi:10.1002/jlac.19707310105.
- ↑ Th. Kauffmann, R. Nürnberg, J. Schulz, R. Stabba: Hetarine, X nachweis eines 5-ring-arins (1-methyl-4.5-dehydroimidazol). In: Tetrahedron. Band 8, Nr. 43, 1967, S. 4273–4280, doi:10.1016/S0040-4039(00)73013-9.
- ↑ Christopher J. Cramer, Stefan Debbert: Heteroatomic substitution in aromatic σ biradicals: the six pyridynes. In: Chem. Phy. Lett. Band 287, Nr. 3–4, 1998, S. 320–326, doi:10.1016/S0009-2614(98)00192-4.
- ↑ Th. Kauffmann, J. Hansen, K. Udluft, R. Wirthwein: Untersuchungen über heterocyclische Arine. In: Angewandte Chemie. Band 76, Nr. 13, 1964, S. 590, doi:10.1002/ange.19640761375.
- ↑ a b c Georg Wittig: 1.2-Dehydrobenzol. In: Angewandte Chemie. Band 77, Nr. 17–18, 1965, S. 752–759, doi:10.1002/ange.19650771704.
- ↑ a b c Rolf Huisgen, H. König: Ringschlußreaktionen über Arine. In: Angewandte Chemie. Band 69, Nr. 8, 1957, S. 268, doi:10.1002/ange.19570690811.
- ↑ a b c d Rolf Huisgen, Jürgen Sauer: Nucleophile aromatische Substitutionen über Arine. In: Angewandte Chemie. Band 72, Nr. 3, 1960, S. 91–108, doi:10.1002/ange.19600720302.
- ↑ Georg Wittig, Reinhard W. Hoffmann: Dehydrobenzol aus 1.2.3-Benzothiadiazol-1.1-dioxyd. In: Chemische Berichte. Band 95, Nr. 11, 1962, S. 2718–2728, doi:10.1002/cber.19620951120.
- ↑ Martin Stiles, Roy G. Miller: DECOMPOSITION OF BENZENEDIAZONIUM-2-CARBOXYLATE. In: J. Am. Chem. Soc. Band 82, Nr. 14, 1960, S. 3802–3802, doi:10.1021/ja01499a094.
- ↑ a b Jan Bülle, Aloys Hüttermann: Das Basiswissen der organischen Chemie: Die wichtigsten organischen Reaktionen im Labor und in der Natur. Georg Thieme Verlag, 2000, ISBN 3-13-119041-8 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Georg Wittig, Hans F. Ebel: über das Auftreten von Dehydrobenzol bei photochemischen und thermischen Prozessen. In: Liebigs Annalen der Chemie. Band 650, Nr. 1, 1961, S. 20–34, doi:10.1002/jlac.19616500103.
- ↑ Ioannis Sapountzis, Wenwei Lin, Markus Fischer, Paul Knochel: Synthese funktionalisierter Arine ausgehend von 2-magnesierten Diarylsulfonaten. In: Angewandte Chemie. Band 116, Nr. 33, 2004, S. 4464–4466, doi:10.1002/ange.200460417.
- ↑ a b Rolf Huisgen, Jürgen Sauer: Nucleophile aromatische Substitutionen, VIII. Kinetik der Freisetzung des Benz-ins aus Halogenbenzolen. In: Chemische Berichte. Volume 92, Nr. 1, 1959, S. 192–202, doi:10.1002/cber.19590920122.
- ↑ R. Huisgen, W. Mack, L. Möbius: Der nachweis der zwischenstufe bei nucleophilen aromatischen substitutionen mit eliminierung; zur struktur der arine. In: Tetrahedron. Band 9, Nr. 1–2, 1960, S. 29–39, doi:10.1016/0040-4020(60)80049-X.
- ↑ Georg Wittig, Liselotte Pohmer: Über das intermediäre Auftreten von Dehydrobenzol. In: Chemische Berichte. Band 89, Nr. 5, 1956, S. 1334–1351, doi:10.1002/cber.19560890539.
- ↑ Rolf Huisgen, Jürgen Sauer: Reaktion der Halogen-naphthaline mit Lithium-piperidid. In: Angewandte Chemie. Band 69, Nr. 11, 1957, S. 390, doi:10.1002/ange.19570691109.
- ↑ George E. Hall, Richard Piccolini, John D. Roberts: Exchange Reactions of Deuterated Benzene Derivatives with Potassium Amide in Liquid Ammonia. In: J. Am. Chem. Soc. Band 77, Nr. 17, 1955, S. 4540–4543, doi:10.1021/ja01622a033.
- ↑ Rolf Huisgen, Wilhelm Mack, Klaus Herbig, Nele Ott, Elisabeth Anneser: Nucleophile aromatische Substitutionen, XIV. Partielle Geschwindigkeitskonstanten der Arin-Bildung aus Bromaromaten mittels Lithiumpiperidids. In: Chemische Berichte. Band 93, Nr. 2, 1960, S. 412–424, doi:10.1002/cber.19600930222.
- ↑ Arine als elektrophile Reagentien, 1. Mitt.: Umsetzung mit Alkylen-phosphoranen. In: Monatshefte für Chemie. Band 75, Nr. 6, 1964, S. 1759–1780, doi:10.1007/BF00901736.
- ↑ John D. Roberts, C. Wheaton Vaughan, L. A. Carlsmith, Dorothy A. Semenow: Orientation in Aminations of Substituted Halobenzenes. In: J. Am. Chem. Soc. Band 78, Nr. 3, 1956, S. 611–614, doi:10.1021/ja01584a025.
- ↑ F. W. Bergstrom, Richard E. Wright, Charles Chandler, W. A. Gilkey: THE ACTION OF BASES ON ORGANIC HALOGEN COMPOUNDS. I. THE REACTION OF ARYL HALIDES WITH POTASSIUM AMIDE. In: J. Org. Chem. Band 1, Nr. 2, 1936, S. 170–178, doi:10.1021/jo01231a007.
- ↑ Albert T. Bottini, John D. Roberts: Mechanisms for Liquid Phase Hydrolyses of Chlorobenzene and Halotoluenes. In: J. Am. Chem. Soc. Band 79, Nr. 6, 1957, S. 1458–1462, doi:10.1021/ja01563a050.
- ↑ Kurt H. Meyer, Friedrich Bergius: Über die Darstellung von Phenol aus Chlor-benzol. In: Berichte der deutschen chemischen Gesellschaft. Band 47, Nr. 3, 1914, S. 3155–3160, doi:10.1002/cber.191404703117.
- ↑ Horst König, Rolf Huisgen: Nucleophile aromatische Substitutionen, XI. Weitere Ringschlußreaktionen über Arine. In: Chemische Berichte. Band 92, Nr. 2, 1959, S. 429–441, doi:10.1002/cber.19590920227.
- ↑ Rolf Huisgen, Horst König, Arthur R. Lepley: Nucleophile aromatische Substitutionen, XVIII. Neue Ringschlüsse über Arine. In: Chemische Berichte. Band 93, Nr. 7, 1960, S. 1496–1506, doi:10.1002/cber.19600930708.
- ↑ S. V. Kessar: Some new aspects of benzyne and radical mediated cyclisations. In: Journal of Chemical Sciences. Band 100, Nr. 2–3, 1988, S. 217–222 (ias.ac.in [PDF]).
- ↑ Bjorn F. Hrutford, J. F. Bunnett: A GENERAL PRINCIPLE FOR THE SYNTHESIS OF HETEROCYCLIC AND HOMOCYCLIC COMPOUNDS. In: J. Am. Chem. Soc. Band 80, Nr. 8, 1958, S. 2021–2022, doi:10.1021/ja01541a065.
- ↑ a b c d Georg Wittig: Bildungsweisen und Reaktionen von Dehydrobenzol (Cyclohexadienin). In: Angewandte Chemie. Band 69, Nr. 8, 1957, S. 245–251, doi:10.1002/ange.19570690802.
- ↑ Georg Wittig, Erhard Knauss: Dehydrobenzol und Cyclopentadien. In: Chemische Berichte. Volume 91, Nr. 5, 1958, S. 895–907, doi:10.1002/cber.19580910502.
- ↑ Georg Wittig, Wolfgang Behnisch: Dehydrobenzol und N-Methyl-pyrrol. In: Chemische Berichte. Volume 91, Nr. 11, 1958, S. 2358–2365, doi:10.1002/cber.19580911116.
- ↑ Martin Stiles, Roy G. Miller: Reaction of Benzyne with Benzene and Naphthalene. In: J. Am. Chem. Soc. Band 85, Nr. 12, 1963, S. 798–1800, doi:10.1021/ja00895a023.
- ↑ Georg Wittig, Renate Ludwig: Triptycen aus Anthracen und Dehydrobenzol. In: Angewandte Chemie. Band 68, Nr. 1, 1956, S. 40, doi:10.1002/ange.19560680107.
- ↑ Harold Hart: Iptycenes, cuppedophanes and cappedophanes. In: Pure & App Chem. Band 65, 1993, S. 27–34, doi:10.1002/jlac.19646730102. Article (PDF; 989 kB)
- ↑ Diego Peña, Sonia Escudero, Dolores Pérez, Enrique Guitián, Luis Castedo: Die erste effiziente Palladium-katalysierte Cyclotrimerisierung von Arinen: Synthese von Triphenylenen. In: Angewandte Chemie. Band 110, Nr. 19, 1998, S. 2804–2806, doi:10.1002/(SICI)1521-3757(19981002)110:19<2804::AID-ANGE2804>3.0.CO;2-2.
- ↑ Enrique Guitián, Dolores Pérez, Diego Peña: Palladium-Catalyzed Cycloaddition Reactions of Arynes. In: Palladium in Organic Synthesis, Topics in Organometallic Chemistry. Band 14, 2005, S. 194–206,, doi:10.1007/b104128.
- ↑ Eiji Yoshikawa, Yoshinori Yamamoto: Kontrollierte Palladium-katalysierte intermolekulare Dehydrobenzol-Dehydrobenzol-Alken- und Dehydrobenzol-Alkin-Alken-Insertion – Synthese von Phenanthren- und Naphthalinderivaten. In: Angewandte Chemie. Band 112, Nr. 1, 2000, S. 185–187, doi:10.1002/(SICI)1521-3757(20000103)112:1<185::AID-ANGE185>3.0.CO;2-N.
- ↑ Diego Peña, Dolores Pérez, Enrique Guitián: Insertion von Arinen in σ-Bindungen. In: Angewandte Chemie. Band 118, Nr. 22, 2006, S. 3659–3661, doi:10.1002/ange.200600291.
- ↑ Ulf Pindur, Ludwig Pfeuffer, Manfred Eitel, Martina Rogge, Manfred Haber: Diels-Alder-Reaktionen von Vinylindolen mit Arin und 1,4-Benzochinonen: Neue potentielle DNA-Interkalatoren. In: Monatshefte für Chemie / Chemical Monthly. Band 122, Nr. 4, 1991, S. 291–298, doi:10.1007/BF00810830.
- ↑ Khalil Shahlai, Samuel Osafo Acquaah, Harold Hart: Use of 1,2,4,5-tetrabromobenzene as a 1,4-benzadiyne equivalent: anti- and syn-1,4,5,8-tetrahydroanthracene 1,4:5,8-diepoxides In: Organic Syntheses. 75, 1978, S. 201, doi:10.15227/orgsyn.075.0201; Coll. Vol. 10, 2004, S. 678 (PDF).
- ↑ a b Rolf Huisgen, Jürgen Sauer, A. Hauser: Katalytische Phenylierung der Chlor-Aromaten mit Phenyl-lithium. In: Angewandte Chemie. Band 69, Nr. 8, 1957, S. 267, doi:10.1002/ange.19570690810.
- ↑ Rolf Huisgen, Jürgen Sauer, Alfred Hauser: Nucleophile aromatische Substitutionen, VI. Katalytische Arylierung der Chloraromaten. In: Chemische Berichte. Volume 91, Nr. 11, 1958, S. 2366–2374, doi:10.1002/cber.19580911117.
- ↑ Rolf Huisgen, Ludwig Zirngibl: Nucleophile aromatische Substitutionen, VII. Sterische und elektronische Faktoren bei der Basen-Addition an 1.2-Naphthin. In: Chemische Berichte. Volume 91, Nr. 11, 1958, S. 2375–2382, doi:10.1002/cber.19580911118.
- ↑ Hiroto Yoshida, Masahiko Watanabe, Joji Ohshita, Atsutaka Kunai: Facile insertion reaction of arynes into carbon–carbon-bonds. In: Chem. Commun. Band 26, 2005, S. 3292–3295, doi:10.1039/B505392G.
- ↑ Hiroto Yoshida, Eiji Shirakawa, Yuki Honda, Tamejiro Hiyama: Addition of Ureas to Arynes: Straightforward Synthesis of Benzodiazepine and Benzodiazocine Derivatives. In: Angewandte Chemie. Band 114, Nr. 17, 2002, S. 3381–3383, doi:10.1002/1521-3757(20020902)114:17<3381::AID-ANGE3381>3.0.CO;2-C.
- ↑ Akkattu T. Biju, Frank Glorius: Eine intermolekulare, durch N-heterocyclische Carbene katalysierte Hydroacylierung von Arinen. In: Angewandte Chemie. Band 122, Nr. 50, 2010, S. 9955–9958, doi:10.1002/ange.201005490.
- ↑ Wenwei Lin, Ioannis Sapountzis, Paul Knochel: Synthese von funktionalisierten Arylmagnesium-Reagentien durch Addition von Magnesiumarylthiolaten und -amiden an Arine. In: Angewandte Chemie. Band 117, Nr. 27, 2005, S. 4330–4333, doi:10.1002/ange.200500443.
- ↑ Adrian L. Smith, K. C. Nicolaou: The Enediyne Antibiotics. In: J. Med. Chem. Band 39, Nr. 11, 1996, S. 2103–2117, doi:10.1021/jm9600398.
- ↑ Marc J. Schottelius, Peter Chen: 9,10-Dehydroanthracene: p-Benzyne-Type Biradicals Abstract Hydrogen Unusually Slowly. In: J. Am. Chem. Soc. Band 118, Nr. 20, 1996, S. 4896–4903, doi:10.1021/ja960181y.
- ↑ a b Samuel J. Danishefsky, Matthew D. Shair: Observations in the Chemistry and Biology of Cyclic Enediyne Antibiotics: Total Syntheses of Calicheamicin γ1 and Dynemicin A. In: J. Org. Chem. Band 61, Nr. 1, 1996, S. 16–44, doi:10.1021/jo951560x.
- ↑ Elfi Kraka, Dieter Cremer: Computer Design of Anticancer Drugs. A New Enediyne Warhead. In: J. Am. Chem. Soc. Band 122, Nr. 34, 2000, S. 8245–8264, doi:10.1021/ja001017k.
- ↑ Chunsong Xie, Yuhong Zhang: A New Tandem Reaction of Benzyne: One-Pot Synthesis of Aryl Amines Containing Anthracene. In: Organic Letters. Band 9, 2007, S. 781–784, doi:10.1021/ol063017g.