Röntgenfluoreszenzanalyse
Die Röntgenfluoreszenzanalyse (RFA), auch Röntgenfluoreszenzspektroskopie (RFS) genannt (englisch X-ray fluorescence spectroscopy, XRF spectroscopy) ist eine Methode aus der Materialanalytik auf Grundlage der Röntgenfluoreszenz. Sie ist eine der am häufigsten eingesetzten Methoden zur qualitativen und quantitativen Bestimmung der elementaren Zusammensetzung einer Probe, da die Proben durch die Messung nicht zerstört werden und keine Aufschlüsse benötigt werden. Besonders breite Anwendung findet sie in der metallverarbeitenden Industrie, bei der Untersuchung von Glas, Keramik und Baustoffen sowie bei der Analyse von Schmierstoffen und Mineralölprodukten. Die Nachweisgrenze liegt etwa bei einem Mikrogramm pro Gramm (ppm).
Sie geht auf Versuche von Richard Glocker (1890–1978) und Hans-Wilhelm Schreiber aus den Jahren 1929 zurück.
Zusammenfassende Beschreibung
[Bearbeiten | Quelltext bearbeiten]Bei der Röntgenfluoreszenzanalyse wird die Technik der Fluoreszenzspektroskopie auf Röntgenstrahlung angewendet. Die Materialprobe wird dabei entweder durch polychromatische Röntgenstrahlung, Gamma- oder Ionenstrahlung angeregt (Anregung mit Elektronenstrahl → EDX). Dabei werden kernnahe Elektronen von inneren Schalen des Atoms herausgeschlagen. Dadurch können Elektronen aus höheren Energieniveaus zurückfallen. Die dabei freiwerdende Energie wird in Form von elementspezifischer Fluoreszenzstrahlung abgegeben. Diese Fluoreszenzstrahlung kann von einem Strahlungsdetektor ausgewertet werden. Die Röntgenfluoreszenzanalyse ermöglicht eine Identifizierung und Konzentrationsbestimmung aller Elemente ab Ordnungszahl Z = 5 (Bor)[1] (also nicht: H (Wasserstoff), He(lium), Li(thium) und Be(ryllium)) in den unterschiedlichsten Zusammensetzungen. Besonders leistungsfähig ist der Nachweis von geringen Verunreinigungen, wie beispielsweise Schwermetallen, die eine hohe Ordnungszahl haben. Das für Stahl wichtigste Legierungselement Kohlenstoff kann jedoch nur mit großem Aufwand nachgewiesen werden.
.jpg)
RFA-Messverfahren
[Bearbeiten | Quelltext bearbeiten]Es existieren bezüglich Anregung und Auswertung verschiedene Verfahren, die für unterschiedliche Einsatzzwecke optimiert sind. Die folgende Aufstellung einiger RFA-Messverfahren kann, auf Grund der zugrunde liegenden Komplexität, leider nie ganz vollständig sein und wird daher stets nur einen Überblick über einige wichtige Verfahren liefern.
Formel zur Quantifizierung:
![{\displaystyle {I_{E_{i,\mathrm {fl} }}(x)=\int _{0}^{x}\mathrm {d} x'I_{E_{0}}\cdot \underbrace {\exp \left[-{\frac {\mu _{i,E_{0}}}{\sin \psi _{\mathrm {in} }}}\ \rho _{i}\ x'\right]} _{\text{Dämpfung der einfallenden Strahlung}}\cdot \underbrace {\left(\tau _{i,E_{0}}\omega _{X_{i}}g_{l,X_{i}}{\frac {j_{X_{i}}-1}{j_{X_{i}}}}\rho _{i}{\frac {1}{\sin \psi _{\mathrm {in} }}}\right)} _{\text{Wahrscheinlichkeit der Photonenentstehung}}\cdot \underbrace {\exp \left[-{\frac {\mu _{i,E_{i,\mathrm {fl} }}}{\sin \psi _{\mathrm {out} }}}\rho _{i}\ x'\right]} _{\text{Dämpfung beim Verlassen der Probe}}\cdot {\frac {\Omega _{\mathrm {Det} }}{4\pi }}\cdot \varepsilon _{\mathrm {Det} ,E_{\mathrm {fl} }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2463cdb2ed551ac8c27e61b97017d2ec45abe78d)

wobei:
- Intensität der messbaren Fluoreszenzstrahlung des Elements mit der Photonenenergie
- Intensität der einfallenden Strahlung mit der Photonenenergie
- Absorptionskoeffizient des Elements bei der Photonenenergie
- Einfallswinkel der anregenden Strahlung
- Dichte des Elements
- photoelektrischer Absorptionsquerschnitt des Elements für ein Photon der Energie









- Fluoreszenz-Ausbeute der Absorptionskante
- Übergangswahrscheinlichkeit der zur Absorptionskante gehörenden Fluoreszenzlinie
- Sprungverhältnis (engl. jump ratio) der Absorptionskante





- Absorptionskoeffizient des Elements bei der Fluoreszenz-Energie ;
- Winkel der Beobachtung / Winkel in dessen Richtung sich der Detektor befindet (oft befindet sich der Detektor senkrecht über der Probe → )
- effektiver Raumwinkel des Detektors
- Effizienz des Detektors bei der Photonenenergie






Hinweis: Im Allgemeinen ist , da die Fluoreszenzstrahlung und nicht der reflektierte Strahl zur Informationsgewinnung verwendet wird. Zwar kann auch mittels des reflektierten Strahls Information über die Probe gewonnen werden. Das ist aber eher Ellipsometrie und nicht explizit Fluoreszenzstrahlung.

Totalreflexions-Röntgenfluoreszenzanalyse (TRFA, TXRF)
[Bearbeiten | Quelltext bearbeiten]Bei der Totalreflexions-Röntgenfluoreszenzanalyse (TRFA, engl. total reflection x-ray fluorescence, TXRF) wird der anregende Röntgenstrahl in einem sehr flachen Einfallswinkel von wenigen Bogenminuten auf die Probe mit Brechungsindex < 1 eingestrahlt, so dass es zu Totalreflexion kommt. Die daraus resultierende Eindringtiefe von wenigen Nanometern erzeugt ein besseres Signal-Rausch-Verhältnis, da Wechselwirkungen mit dem Trägermaterial der Probe nicht stattfinden. So kann die Nachweisgrenze bis auf 0,01 Pikogramm erweitert werden. Des Weiteren bilden sich durch Wechselwirkung der einfallenden mit den reflektierten Röntgenstrahlen stehende Wellenfelder (engl. x-ray standing waves, XSW) aus, die insbesondere zur Feststellung und Quantifizierung von Verunreinigungen auf der Probe (z. B. einem Wafer) verwendet werden können.[2]
Röntgenfluoreszenzanalyse unter streifendem Einfall (GIXRF)
[Bearbeiten | Quelltext bearbeiten]Hierbei handelt es sich um Röntgenfluoreszenzanalyse unter streifendem Einfall (englisch grazing-incidence x-ray fluorescence, GIXRF). Dieses Verfahren ist eng verwandt mit TXRF, unterscheidet sich jedoch unter anderem dahingehend, dass bei GIXRF der Einfallswinkel um den Bereich des kritischen Winkels der Totalreflexion variiert wird. Unterhalb des kritischen Winkels handelt es sich also prinzipiell um das klassische TXRF-Verfahren, überschreitet der Einfallswinkel jedoch den kritischen Winkel so dringt die Strahlung mit zunehmendem Winkel in die darunter liegende Schicht ein. Des Weiteren bilden sich auch bei GIXRF die von TXRF bekannten stehenden Wellenfelder der Röntgenstrahlung (x-ray standing waves, XSW) aus, wodurch es zu einer Modulation der Anregungsstrahlung im Bereich der Grenzfläche kommt. Dadurch wird es möglich zusätzlich etwas über die Beschaffenheit der Grenzschicht zu lernen bzw. die Elementzusammensetzung einer Schicht oder Probe tiefenabhängig zu bestimmen[3].

Dieses Verfahren wird bei Schichtsystemen eingesetzt deren Schichten sich im Brechungsindex deutlich unterscheiden. Dies trifft unter anderem auf Solarzellen zu. Durch den unterschiedlichen Brechungsindex unterscheiden sich auch die kritischen Winkel der einzelnen Schichten. Wenn der kritische Winkel einer oberen Schicht größer ist, als der kritische Winkel einer darunterliegenden Schicht, dann gibt es einen Winkelbereich, in dem das Röntgenlicht die obere Schicht durchdringt, die darunter liegende Schicht aber auf Grund von Totalreflexion nicht erreicht. Ein großer Teil der totalreflektierten Strahlung überwindet anschließend die Grenze der Probe und tritt aus der oberen Schicht wieder aus. Dort überlagert sie sich mit dem Teil der Strahlung, der an der obersten Oberfläche der Probe reflektiert wurde. Aus der sich ergebenden Interferenz im gesamten reflektierten Röntgenlicht kann man auf die Dicke der durchstrahlten Schicht schließen.


Mikroröntgenfluoreszenzanalyse (μ-RFA, μ-XRF)
[Bearbeiten | Quelltext bearbeiten]Bei der Mikroröntgenfluoreszenzanalyse (μ-RFA, engl. micro x-ray fluorescence, μ-XRF) handelt es sich um ein Verfahren, bei dem der Röntgenstrahl mittels Röntgenoptiken auf wenige Mikrometer fokussiert wird, um somit eine Auflösung im Mikrometer-Bereich zu erhalten.
Eine erst in den 2000er Jahren an der TU Berlin entwickelte Erweiterung dieses Verfahrens zur 3D-Mikroröntgenfluoreszenzspektroskopie (3D-μ-RFA, 3D-μ-XRF) erlaubt es Proben dreidimensional (3D) zerstörungsfrei abzurastern. Dabei wird sowohl der Anregungs- als auch der Detektionsstrahl durch je eine Röntgenlinse (sogenannte Polykapillarlinse) geleitet, wodurch ein nur wenige Kubikmikrometer messendes Untersuchungsvolumen definiert wird.[4]

Röntgenabsorptionsspektroskopie (XAS)
[Bearbeiten | Quelltext bearbeiten]Eine weitere Gruppe von Untersuchungsverfahren stellt die Röntgenabsorptionsspektroskopie (engl. X-ray absorption spectroscopy, XAS) dar. Sie umfasst Methoden, bei denen die Absorption von Röntgenstrahlung im Bereich einer Absorptionskante gemessen wird. Da in der Regel die Feinstruktur des erhaltenen Spektrums analysiert wird, bezeichnet man diese Methodengruppe auch als Röntgenabsorptions-Feinstruktur-Spektroskopie (engl. X-ray absorption fine structure spectroscopy, XAFS spectroscopy). Grundsätzlich unterteilt man diese Gruppe in die Untersuchung der „Nahkante“ (NEXAFS / XANES) und die Untersuchung der „erweiterten Kantenstruktur“ (EXAFS) auf.
Bei der Röntgen-Nahkanten-Feinstruktur-Spektroskopie (engl. near-edge x-ray absorption fine structure, NEXAFS) bzw. (X-ray absorption near-edge structure, XANES) handelt es sich um ein Verfahren, das die Absorptionskante eines Elements hoch aufgelöst untersucht, wodurch die Bezeichnung Absorption begründet ist. Gelegentlich führt dieses zu der nicht ganz richtigen Annahme, dass es sich um reine Absorptionsspektroskopie handelt.
Bei der EXAFS-Spektroskopie (von engl. extended X-ray absorption fine structure, EXAFS) handelt es sich um eine Methode die den „erweiterten Bereich“ der Absorptionskante betrachtet, d. h. die kantenferne Röntgenabsorptionsfeinstruktur. Bei der Untersuchung von Molekülen an Oberflächen wird diese Methode auch als SEXAFS (engl. surface extended X-ray absorption fine structure) bezeichnet.
In beiden Fällen kommt es zu Interferenzen (Pfeile), die die Grundlage für quantitative Untersuchungen bei NEXAFS/XANES- bzw. EXAFS-Messungen liefern. Zum Beispiel lässt sich die Frequenz der Oszillationen auf die Abstände zu den Nachbaratomen zurückführen. Beide Verfahren werden sowohl im „Absorptionsmodus“, also in Transmission, als auch im „Fluoreszenzmodus“ verwendet. Da aber nur dünne Probensysteme überhaupt transmittierend, also durchlässig sind, wird dieses Verfahren meistens im Fluoreszenzmodus betrieben und gehört somit ebenfalls zur Gruppe der Röntgenfluoreszenzanalyse (RFA).
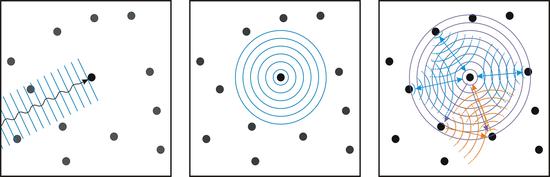
Links: Die einfallende ebene Welle wird von einem Atom absorbiert. Mitte: Das absorbierende Atom emittiert seinerseits eine sphärische Photoelektronenwelle. Rechts: Die emittierte Photoelektronenwelle wird an umliegenden Atomen gestreut. Es kommt dabei einerseits (blau) zu Einfachstreuprozessen die die Ursache für die EXAFS-Strukturen sind, andererseits (orange) kommt es aber auch zu Mehrfachstreuprozessen die die Ursache für die NEXAFS/XANES-Strukturen sind.
Detektionsarten
[Bearbeiten | Quelltext bearbeiten]Bei der RFA gilt es grundsätzlich zwei Arten der Detektion zu unterscheiden
- Die Energie-dispersive Detektion der Fluoreszenz (EDXRF) und
- die Wellenlängen-dispersive Detektion der Fluoreszenz (WDXRF).
Energiedispersive Röntgenfluoreszenzanalyse (EDXRF)
[Bearbeiten | Quelltext bearbeiten]Bei der energiedispersiven Röntgenfluoreszenzanalyse (EDRFA, engl. energy dispersive X-Ray fluorescence, EDXRF) wird die Anregung der Probe durch Röntgenstrahlen erreicht. Zur Anregung bestimmter, gewünschter Elemente oder zur Unterdrückung von Hintergrundrauschen können Filter aus verschiedenen Elementen zwischen die Röntgenquelle und die Probe geschaltet werden. Ein energiedispersiver Detektor misst, ähnlich wie bei der EDX, die Energie der ausgestrahlten Fluoreszenzquanten.
Wellenlängendispersive Röntgenfluoreszenzanalyse (WDXRF)
[Bearbeiten | Quelltext bearbeiten]Bei der wellenlängendispersiven Röntgenfluoreszenzanalyse (WDRFA, engl. wavelength dispersive X-Ray fluorescence, WDXRF) erfolgt die Anregung genau wie beim EDXRF. Der Unterschied liegt in der Detektion und Auswertung der emittierten Fluoreszenzstrahlung: Diese werden durch einen Kollimator parallel ausgerichtet, in einem Analysatorkristall gebeugt und durch einen geeigneten Detektor registriert. Der Kristall dient dabei dazu, durch Beugung das Spektrum der von der Probe ausgehenden polychromatischen Sekundärstrahlung nach Wellenlängen aufzuspalten und anhand des Beugungswinkels der Röntgenstrahlung die qualitative Bestimmung des Elementes und durch Messung der Intensität der Röntgenstrahlung eine quantitative Bestimmung zu ermöglichen.
Vergleich EDXRF/WDXRF
[Bearbeiten | Quelltext bearbeiten]Da die Wellenlänge eines Röntgenquants indirekt proportional zu seiner Energie ist, wäre zu erwarten, dass die Ergebnisse von EDXRF und WDXRF bis auf eine Spiegelung des Spektrums identisch wären. Tatsächlich ergeben sich aber aufgrund der unterschiedlichen Bauart einige signifikante Unterschiede:
Die Energieauflösung beschreibt die Trennschärfe zweier spektraler Peaks. Sie wird meist für die Röntgenenergie von 5,9 keV (Mangan-K-alpha-Linie, Mn-K-α-Linie) angegeben. Die Auflösung eines WDXRF-Systems hängt vom Kristall und dem Design der Optik ab. Es können Auflösungen von 20 eV bis 5 eV erreicht werden. Dagegen erreicht die Auflösung eines EDXRF-Systems nur Werte von 600 eV bis 120 eV. Damit ist ein WDXRF-System deutlich genauer, so dass auch nahe beieinander liegende Peaks noch getrennt werden können. Allerdings sind die hochgenauen Kristalle und Optiken teuer und fehleranfällig. Zudem erfordern WDXRF-Systeme deutlich längere Messzeiten.
Die Effizienz beschreibt, wie gut die Röntgenstrahlung der Röntgenquelle genutzt wird, um die Probe anzuregen und dort Röntgenstrahlung zu emittieren. Dieser Faktor bestimmt wesentlich, welche Leistung die Röntgenquelle haben muss und ist damit einer der zentralen Kostenfaktoren. Das WDXRF ist hier deutlich im Nachteil, da beim EDXRF mit direkter Anregung so gut wie keine Energie verloren geht, wohingegen beim WDXRF fast die hundertfache Leistung eingesetzt werden muss, um die gleiche Ausbeute an Röntgenquanten zu erreichen.
Das EDXRF stellt eine deutlich kostengünstigere Variante dar, die allerdings auch eine deutlich geringere Energieauflösung bietet, so dass je nach Anwendung entschieden werden muss, welche Bauform geeigneter ist.
Komponenten
[Bearbeiten | Quelltext bearbeiten]Strahlungsquelle
[Bearbeiten | Quelltext bearbeiten]Als Strahlungsquelle können folgende Instrumente eingesetzt werden
- eine Röntgenröhre.
- Seitenfensterröhre. Hierbei wird eine Anode aus Chrom, Wolfram, Molybdän, Gold oder Rhodium mit einem Elektronenstrahl beschossen. Es entsteht sehr viel Wärme und Röntgenstrahlung, die die Röntgenröhre durch die Beryllium-Fenster an den Seiten verlässt.
- Weitaus häufiger wird wegen der besseren Strahlendichte eine Endfensterröhre eingesetzt. Hierbei befindet sich die Anode gegenüber vom Beryllium–Fenster, und die Kathode ist ringförmig um die Anode aufgebaut. Legt man eine Spannung an, wandern die Elektronen auf einer gebogenen Bahn zur Anode.
- Radioaktive Nuklide. Für transportable Spektrometer können statt einer Röntgenröhre auch Primärstrahler wie Eisen (55Fe) oder Plutonium (238Pu) verwendet werden. Allerdings sind die Nachweisgrenzen hier sehr hoch.
- eine Synchrotronstrahlungsquelle.
Filter
[Bearbeiten | Quelltext bearbeiten]Verwendet man eine Röntgenröhre als Strahlungsquelle, besteht die erzeugte Röntgenstrahlung zum einen aus Bremsstrahlung und zum anderen aus einem charakteristischen Linienspektrum des beschossenen Anodenmaterials. Wird beispielsweise Chrom als Anodenmaterial verwendet, wird man auch das charakteristische Linienspektrum von Chrom am Ende detektieren. Es kann dabei nicht unterschieden werden, ob das Linienspektrum nur aus der Röntgenröhre stammt oder ob die Probe auch noch Chrom enthält. Daher wird zwischen Röhre und Probe ein Selektivfilter gesetzt, um die charakteristischen Kbeta- und Kalpha-Linien zu absorbieren. Das Material des Selektivfilters wird so gewählt, dass dessen Ordnungszahl um eins oder zwei kleiner ist als das Element, aus dem die Anode besteht. Beispielsweise wird ein Titanfilter (Ordnungszahl 22) für eine Chromröhre (Ordnungszahl 24) verwendet.
Spaltsystem
[Bearbeiten | Quelltext bearbeiten]Als Spaltsystem können sowohl dünne Rohre (Kollimatoren) als auch Metall-Lamellen (Soller-Blenden) verwendet werden. Ihr Zweck ist es, aus der divergenten Strahlung ein paralleles Bündel zu selektieren.
Analysatorkristall
[Bearbeiten | Quelltext bearbeiten]Um die Fluoreszenzlinien der Röntgenstrahlung später analysieren zu können, muss sie zunächst an einem regelmäßigen Gitter gebeugt werden. Als Beugungsgitter für Röntgenstrahlung bieten sich Einkristalle wie zum Beispiel LiF-Einkristalle oder Multilagenspiegel an. Bragg-Gleichung

wobei:
- n die Beugungsordnung;
- d den Gitterabstand;
- θ den Glanz- oder Braggwinkel;
- λ die Wellenlänge bezeichnen. Die längste messbare Wellenlänge λmax ergibt sich durch Einsetzen von θmax = 90°.
Korrigierte Bragg-Gleichung für Multilagenspiegel:

wobei:
- δ die Dispersion der beteiligten Schichtmaterialien
Szintillationszähler
[Bearbeiten | Quelltext bearbeiten]Szintillationszähler werden für Elemente mit einer höheren Ordnungszahl als Eisen (26 Protonen) verwendet und bestehen meist aus einem NaI–Kristall, welcher mit Thallium dotiert ist. Trifft die Röntgenstrahlung auf den Kristall, wird die Röntgenstrahlung in fluoreszierende Strahlung umgewandelt. Die fluoreszierende Strahlung wird in dem nachgeschalteten Photomultiplier in elektrische Impulse verwandelt und um ein Vielfaches verstärkt.
Zählrohr
[Bearbeiten | Quelltext bearbeiten]Zählrohre werden zur Messung von längerwelliger Strahlung eingesetzt, welche von den leichteren Elementen Beryllium (4 Protonen) bis Mangan (25 Protonen) ausgesendet wird. Ein Zählrohr ist mit einem Inertgas (beispielsweise Argon) gefüllt. Trifft Röntgenstrahlung auf ein Argonatom, schlägt es ein Photoelektron heraus. Dieses Photoelektron wandert zur Drahtanode und erzeugt auf dem Weg dorthin durch Sekundär-Stoßionisation bis zu 10.000 Elektron-Ion-Paare (Gasverstärkung). Die Rückwanderung der positiven Ionen zur Zählerwand verursacht eine kurzzeitige (Mikrosekunde) Störung des elektrischen Feldes, was dann am Vorverstärker einen Strom-/Spannungsimpuls erzeugt. Die Höhe dieses Impulses ist proportional zur eingestrahlten Energie des Röntgenquants (vgl. Proportionalzähler – im Gegensatz zum Geigerzähler, in dem die Information über die Energie verloren geht).
Anwendung in der Praxis
[Bearbeiten | Quelltext bearbeiten]Grenzen der Methode
[Bearbeiten | Quelltext bearbeiten]Die Röntgenfluoreszenzanalyse kann nicht auf Elemente angewendet werden, die leichter als Bor (Ordnungszahl 5) sind. Vernünftige Analysenwerte sind erst ab Fluor (Ordnungszahl 9) (dazwischen liegen: C (Kohlenstoff), N (Stickstoff), und O (Sauerstoff)), gute Werte erst ab Natrium (Ordnungszahl 11) (dazwischen Ne(on) (Ordnungszahl 10)) möglich, da die Röntgenstrahlung der leichteren Elemente so leicht absorbiert wird, dass sie gar nicht erst in den Detektor eindringen kann. Die quantitative Obergrenze ergibt sich nach den jeweiligen Referenzproben (siehe Kalibrierung).
Art und Form der zu analysierenden Probe
[Bearbeiten | Quelltext bearbeiten]Im Allgemeinen analysiert man feste Proben. Flüssigkeiten werden in einem Plastik-Gefäß mit einem Boden aus einer dünnen Folie analysiert. Meist verwendet man feste Probenkörper, die die Form einer runden Scheibe (ähnlich einem großen Geldstück) mit einem Durchmesser von 2 bis 5 cm haben. Die Probe muss mindestens eine ebene Fläche haben, von der die Röntgenstrahlen reflektiert werden können.
Mit leicht tragbaren „handgehaltenen RFA-Spektrometern“ können in Sekundenschnelle vor Ort Elementanalysen oder Spurenelementanalysen beispielsweise bei Metallen (Neuware, Abfälle, zur Sortierung, bei Kontrollen), Kunststoffen, Nahrungsmitteln, Beschichtungen, Erze und Mineralien im Bergbau und bei der Exploration etc. ermittelt werden, ohne dafür eine Probe an ein Chemielabor senden zu müssen.
Probenpräparation
[Bearbeiten | Quelltext bearbeiten]Am einfachsten kann man Metallscheiben analysieren. Pulverförmige Proben müssen erst fein gemahlen und zusammen mit einem Bindemittel (beispielsweise Paraffinwachs oder Cellulosepulver) zu einer Probentablette gepresst werden. Eine andere Möglichkeit ist das Mischen von Gesteinspulver etc. mit Lithiumtetraborat und die Herstellung einer glasartigen Schmelze, welche in eine Gießform gegossen wird. Bei diesem Vorgang wird die Probe natürlich zerstört.
Pulvertabletten für die Analyse von Gesteinspulvern, Zement, Schlacke, Flugasche
[Bearbeiten | Quelltext bearbeiten]3 Gramm der Untersuchungssubstanz werden mit 0,6 Gramm Paraffinwachs-Pulver gemischt und in einer Tablettenpresse gepresst. Stabilere Tabletten werden mit 2 Gramm Borsäure erreicht, wenn die Probenmischung aufgegeben und daraus eine Tablette gepresst wird. So befindet sich die Substanz auf der Borsäureschicht, die unter Druck bessere Fließeigenschaften hat.
Schmelztabletten für die Analyse von Gesteinspulvern, Zement, Schlacke
[Bearbeiten | Quelltext bearbeiten]Bessere Messergebnisse werden mit Schmelztabletten erreicht. Dazu wird ein Gewichtsteil von 1 bis 2 Gramm des Gesteinspulvers und fünf Teilen von 5 bis 10 Gramm Dilithiumtetraborat (Li2B4O7) im Achatmörser gründlich verrieben und dadurch vermischt, anschließend in einen Platin-Tiegel gegeben. Der Inhalt im Tiegel wird im Elektroofen mindestens 12 Minuten auf 1050 bis 1080 °C erhitzt und die flüssige Schmelze in eine Gießform über dem Bunsenbrenner überführt. Diese Schmelze in der Gießform wird mit Pressluft abgekühlt, damit die Schmelze zwischen 1000 °C und 600 °C nicht auskristallisiert. Zemente werden nur bis zur schwachen Rotglut gekühlt, danach wird langsamer ausgekühlt um das Zerspringen der Tabletten zu verhindern. Gegen das Ankleben der Tablette an der Gießform, wodurch die Tablette zerspringt, wird zwei Minuten vor dem Abgießen eine kleine Menge Lithiumjodid (20 mg) zugegeben. Bei wenig Probensubstanz können auch 250 mg Probe mit 7,25 g Lithiumtetraborat angesetzt werden.
Beim Gießen der Tabletten sind Ofenschaubrille, lange Tiegelzange und Schutzhandschuhe nötig, um vor der Wärmestrahlung zu schützen. Beim Zugeben von Lithiumjodid entstehen Ioddämpfe, daher sollte der Ofen unter einem Abzug stehen.
Für Schmelztabletten existieren auch automatische Aufschlussgeräte, die bis zu 6 Schmelzaufschlüsse gleichzeitig produzieren.
Nachweisgrenzen in Schmelztabletten
[Bearbeiten | Quelltext bearbeiten]Es wird ein Mischungsverhältnis von 1 + 59 zugrunde gelegt. Erreichbare Mindest-Nachweisgrenzen liegen bei wenigstens:
- Natrium, Magnesium: 100 ppm
- Aluminium, Silicium, Phosphor, Schwefel: 50 ppm
- Kalium, Calcium, Barium, Titan: 30 ppm
- Eisen, Mangan, Chrom, Nickel, Kupfer, Zinn: 10 ppm
- Rubidium, Strontium, Yttrium, Zirconium, Niob: 6 ppm
Die Nachweisgrenze von Pulvertabletten ist um den Faktor 2 bis 3 besser, auf Grund von Korngrößeneffekten und schlechterer Homogenität der Proben ist aber die Genauigkeit der Analysen geringer, die Toleranzen sind größer.
Kalibrierung des Röntgenfluoreszenzgerätes
[Bearbeiten | Quelltext bearbeiten]Man verwendet Kalibrierproben bekannten Gehaltes, die man sich entweder selbst herstellt oder käufliche Standardproben, deren Gehalte von vielen renommierten Labors ermittelt wurden. Drei Kalibrierverfahren sind Kalibrierung über externe Standards, interne Standards oder Standardaddition, wobei in der Röntgenfluoreszenzanalyse meistens die Methode des internen Standards Verwendung findet. Mittlerweile gibt es auch Instrumente, die (bei geringerer Genauigkeit) auch ohne Kalibrierproben auskommen.[5]
Analyse beim Goldankauf
[Bearbeiten | Quelltext bearbeiten]Durch den stetigen Anstieg des Goldpreises und den damit wachsenden Markt für Edelmetalle findet die RF-Analyse auch im Bereich des Goldankaufs immer mehr Verwendung. Der Feingehalt der Probe kann hier im Gegensatz zur herkömmlichen Strich/Säure Methode zerstörungsfrei analysiert werden. Proben unbekannter Zusammensetzung, mit einer großen Zahl von Begleitelementen, können so korrekt bestimmt werden. Somit werden auch betrügerische Objekte mit gefälschtem Feingehaltstempel nachgewiesen. Damit bietet die RF-Analyse gegenüber herkömmlichen Analysen mehr Transparenz, hat jedoch leider den Nachteil einer nur oberflächlichen Bestimmung der Zusammensetzung. Eine genaue Analyse in die Tiefe einer Probe ist nicht möglich. Einem möglichen Betrug sind auch hier keine Schranken gesetzt. Für eine korrekte Analyse ist eine Zerstörung der Probe unumgänglich. Eine zerstörungsfreie Goldanalyse ist dagegen über die Messung der Ultraschallgeschwindigkeit möglich.[6]
Analyse von Kunstwerken
[Bearbeiten | Quelltext bearbeiten]Die RF-Analyse wurde mit Erfolg auch bei der Analyse von Gemälden angewendet.[7] In der letzten Zeit sind miniaturisierte Analysengeräte verfügbar geworden, welche eine Analyse vor Ort erlauben. Beim Einsatz größerer Scan-Analysengeräte kann die Verteilung einzelner chemischer Elemente im Gemälde abgebildet und somit auch die eingesetzten Pigmente identifiziert werden. Bei der Untersuchung von Rembrandts Porträt eines Mannes im Militärkostüm[8] wurde mittels RFA und Neutronenaktivierungsanalyse ein darunterliegendes übermaltes Porträt eines Mannes entdeckt. Ein übermaltes Frauenporträt konnte auch bei der Untersuchung des Bildes „Grass“ von Vincent van Gogh unter den Oberflächenschichten sichtbar gemacht werden.[9]
Online-Analyse und Online-Separation von Wertstoffen
[Bearbeiten | Quelltext bearbeiten]Die energiedispersive RF-Analyse bietet sich an, eine zerstörungsfreie Analyse von Wertstoffen durchzuführen, mit dem Ziel, diese sortenrein für Recyclingzwecke zu trennen. Dabei sind Verunreinigungen der Oberfläche, Lackanhaftungen und ähnliches von geringem Einfluss auf das Messergebnis, da diese von der anregenden und fluoreszierenden Röntgenstrahlung durchdrungen werden. Das Verfahren wurde erstmals 1992 der Öffentlichkeit vorgestellt, indem geschredderter Automobilschrott (Partikelgröße 3 bis 5 cm) in einem Online-Verfahren nach unterschiedlichen Metallen vollautomatisch sortiert wurde. Zum Einsatz kamen als Anregung eine radioaktive Strahlenquelle (Americium 241), ein mit flüssigem Stickstoff gekühlter Halbleiterdetektor und eine ultraschnelle Messeinheit bestehend aus Signalaufbereitung, Analog-Digital-Wandlung und einem Vielkanalanalysator. Die sich auf einem Transportband befindlichen, mechanisch separierten Schrottteile ("Perlenschnur-Aufreihung") passieren zunächst die Detektionseinheit. Während des Weitertransports findet die rechnerseitige Auswertung, Analyse und Auswertung statt, so dass entsprechende seitlich angebrachte Auswurfeinheiten, die jeweils eine Metallsorte repräsentierten, getriggert werden können. Per Druckluftstoß werden die Schrottteile in den der Metallsorte entsprechenden Auswurfbehälter vom Transportband geschossen. Die Vollanalyse eines einzelnen Schrottteils wurde in 10 ms realisiert, so dass mit dem Verfahren ein Durchsatz von 1000 kg pro Stunde realisiert werden konnte. Das Verfahren wurde so weiter entwickelt, dass es in der Lage war, Bildschirmglasbruch von Röhrenfernsehern und Röhrenmonitoren (Frontglas versus Rückglas) sortenfrei zu sortieren. Im Vergleich zu Röntgenröhren als Anregung hat sich gezeigt, dass der Einsatz einer Feststoff-Strahlenquelle deutlich robuster ist, zumal die strahlenschutzrechtlichen Anforderungen und Auflagen nur minimale Unterschiede aufweisen.[10][11]
Literatur
[Bearbeiten | Quelltext bearbeiten]- R. Tertian, F. Claisse: Principles of quantitative X-ray fluorescence analysis. Heyden & Son, 1982, ISBN 0-85501-709-0.
- B. K. Agarwal: X-Ray Spectroscopy: An Introduction. Springer, 1991, ISBN 0-387-09268-4.
- R. Klockenkaemper: Total-Reflection X-Ray Fluorescence Analysis. John Wiley & Sons, 1996, ISBN 0-471-30524-3.
- R. Van Grieken, A. A. Markowicz: Handbook of X-Ray Spectrometry. Marcel Dekker, 2002, ISBN 0-8247-0600-5.
- B. Beckhoff u. a.: Handbook of Practical X-Ray Fluorescence Analysis. Springer, 2006, ISBN 3-540-28603-9.
- Georg Schwedt: Analytische Chemie: Grundlagen, Methoden und Praxis. Wiley-VCH, 2008, ISBN 978-3-527-31206-1.
- H. Erhardt: Röntgenfluoreszenzanalyse, Anwendungen in Betriebslaboratorien. Deutscher Verlag für Grundstoffindustrie, Leipzig 1988, ISBN 3-342-00219-0.
- Paula Hahn-Weinheimer, Klaus Weber-Diefenbach, Alfred Hirner: Röntgenfluoreszenzanalytische Methoden, Grundlagen und praktische Anwendung in den Geo-, Material- und Umweltwissenschaften. Springer-Verlag, 2000, ISBN 3-528-06579-6.
Weblinks
[Bearbeiten | Quelltext bearbeiten]Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ D. A. Skoog, J. J. Leary: Instrumentelle Analytik Grundlagen – Geräte – Anwendung 1992. Springer Verlag, Berlin, S. 410.
- ↑ Burkhard Beckhoff, Rolf Fliegauf, Michael Kolbe, Matthias Müller, Jan Weser, Gerhard Ulm: Reference-Free Total Reflection X-ray Fluorescence Analysis of Semiconductor Surfaces with Synchrotron Radiation. In: Analytical Chemistry. Band 79, Nr. 20, 1. September 2007, S. 7873–7882, doi:10.1021/ac071236p.
- ↑ P. Hönicke, B. Beckhoff, M. Kolbe, D. Giubertoni, J. van den Berg, G. Pepponi: Depth profile characterization of ultra shallow junction implants. In: Analytical Bioanalytical Chemistry. Band 396, Nr. 8, 1. April 2010, S. 2825–2832, doi:10.1007/s00216-009-3266-y.
- ↑ Ioanna Mantouvalou: Quantitative 3D Micro X-ray fluorescence spectroscopy. 2009, urn:nbn:de:kobv:83-opus-23153 (Dissertationsarbeit, Technische Universität Berlin, 2009).
- ↑ Introduction of UniQuant®. Abgerufen am 9. Januar 2022.
- ↑ René Böttcher, Lothar Spieß: Bestimmung von Werkstoffkennwerten mit Ultraschallverfahren – ist der „Goldbarren“ echt? In: ZfP in Forschung, Entwicklung und Anwendung / DGZfP-Jahrestagung Zerstörungsfreie Materialprüfung; 2014 (Potsdam): 26–28. Mai 2014. DGZfP, Berlin 2014 (7 Seiten).(PDF-Datei), abgerufen am 9. Januar 2022
- ↑ X-Ray Fluorescence. Description of X-Ray Fluorescence. ColourLex, abgerufen am 9. Januar 2022.
- ↑ Karen Trentelman, Koen Janssens, Geert van der Snickt, Yvonne Szafran, Anne T. Woollett, Joris Dik: Rembrandt’s An Old Man in Military Costume: the underlying image re-examined. In: Applied Physics A. November 2015, Band 121, Nr. 3, S. 801–811, DOI:10.1007/s00339-015-9426-3.
- ↑ Joris Dik, Koen Janssens, Geert Van Der Snickt, Luuk van der Loeff, Karen Rickers, Marine Cotte: Visualization of a Lost Painting by Vincent van Gogh Using Synchrotron Radiation Based X-ray Fluorescence Elemental Mapping. In: Analytical Chemistry. Band 80, Nr. 16, August 2008, S. 6436–6442, doi:10.1021/ac800965g.
- ↑ Martin Przewloka, Burkhard Poppeck: Verfahren zur kontinuierlichen Bestimmung der Zusammensetzung eines Materialstromes und dafür geeignete Vorrichtung. In: Patent (Hrsg.): Deutsches Patentamt. WO1999046584A3, 10. März 1999.
- ↑ Zeitung für Rheinberg und Xanten: Blitzschnell wird der Schrott "enttarnt". Nummer: 125, 28. Mai 1992.