Tyrosinase
| Tyrosinase | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 511 Aminosäuren | |
| Sekundär- bis Quartärstruktur | Membranprotein | |
| Kofaktor | 2 Kupfer | |
| Isoformen | 2 | |
| Bezeichner | ||
| Gen-Name | TYR | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 1.14.18.1, Dioxygenase | |
| Reaktionsart | Oxidation | |
| Substrat | L-Tyrosin + L-Dopa + O2 | |
| Produkte | L-Dopa + Dopachinon + H2O | |
| Vorkommen | ||
| Homologie-Familie | Tyrosinase | |
| Übergeordnetes Taxon | Lebewesen | |
Tyrosinase ist ein Kupfer enthaltendes Enzym, das die Oxidation von Phenolen, z. B. Tyrosin, katalysiert. Es ist weit verbreitet in fast allen Lebewesen.
Bei Tieren und Menschen ist Tyrosinase (zusammen mit den Enzymen Tyrp1 und Dct) an der Synthese von Melanin an der Membran von Melanozyten beteiligt und daher unentbehrlich für den Schutz vor UV-Strahlung. Bei einem Teil der Organismen mit Albinismus ist das Enzym verändert oder fehlt ganz. Die Tyrosinase in Pflanzen hat zusätzliche Funktionen und heißt Polyphenoloxidase.
Melanogenese
[Bearbeiten | Quelltext bearbeiten]Die Produktion von Tyrosinase steigt bei erhöhter UVB-Strahlung. Dann laufen an der Zellmembran von Melanoblasten folgende chemische Reaktionen verstärkt ab.[1]
 + 1/2 O2 →
+ 1/2 O2 → 


Tyrosin wird zunächst zu L-Dopa oxidiert.
- + 1/2 O2 →
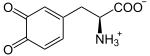 + H2O
+ H2O

L-Dopa wird weiteroxidiert zu Dopachinon, welches sich eigenständig in zwei Schritten zu Dopachrom oxidiert. Von hier ist ein Reaktionsweg in zwei Schritten mithilfe der Enzyme TYRP1 und DCT zum Melanin möglich. Die Tyrosinase kann aber auch selbst Melanin aus Dopachrom herstellen:
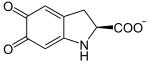 →
→  + CO2
+ CO2


Dopachrom wird zu 5,6-Dihydroxyindol (DHI) decarboxyliert und umgelagert.
- + O2 →
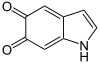 + 2 H2O
+ 2 H2O

DHI wird zu Indol-5,6-chinon oxidiert, welches mithilfe Sauerstoff letztendlich zu Melanin polymerisiert (nicht gezeigt).
Daraus folgt, dass Tyrosinase unbedingt erforderlich für die Melaninsynthese ist, während ein Mangel an Tyrp1 oder Dct durch die Tyrosinase mehr oder weniger ausgeglichen werden kann. Die Menge erzeugten Melanins ist in jedem Fall reduziert, so dass hier eine einfache Erklärung für genetische Variationen in der Farbe von Haut, Augen und Haaren liegt.[1][2]
ortho-Vanillin ist ein schwacher und Monobenzon ein starker Inhibitor der Tyrosinase.[3][4]
Pathologie
[Bearbeiten | Quelltext bearbeiten]Mutationen am TYR-Gen sind für den Okulokutanen Albinismus Typ 1 (OCA1) verantwortlich.[5] Eine Variante des Waardenburg-Syndroms Typ 2 entsteht durch eine Interaktion einer Mutation des Tyrosinaselocus (R402Q) mit einer Mutation des Mitf-Gens.[6]
Albinismus ist allgemein mit ophthalmologischen Besonderheiten verknüpft, wie Nystagmus, Strabismus, starke refraktive Fehler, foveale Dysgenese, chorioretinale Hypopigmentation. Alle Säugetiere, von denen ein OCA1-Defekt bekannt ist, weisen bereits bei Vorhandenseins auch nur eines OCA1-Allels Defekte im visuellen System auf, die nicht durch die Menge des produzierten Melanins erklärt werden können, und deren genaue Ursache noch unbekannt ist. Bei einem dieser Defekte, einer Dysgenese der Fovea centralis, ist keine foveale Einsenkung vorhanden, vielmehr ist die Netzhaut in diesem Bereich so dick wie überall sonst (300 µm gegenüber 150 µm). Da geschätzte ein bis zwei Prozent der Menschen heterozygot eine Mutation in TYR tragen, wird vermutet, dass unerklärte Fälle von Stereoblindheit darauf zurückzuführen sind.[7][8][9]
Weblinks
[Bearbeiten | Quelltext bearbeiten]Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ a b UniProt P14679.
- ↑ Skin/Hair/Eye pigmentation variation 3. In: Online Mendelian Inheritance in Man. (englisch).
- ↑ Isao Kubo, Ikuyo Kinst-Hori: Tyrosinase inhibitory activity of the olive oil flavor compounds, in: Journal of Agricultural and Food Chemistry, 47 (11), 1999, S. 4574–4578; doi:10.1021/jf990165v.
- ↑ Franz v. Bruchhausen, G. Dannhardt, Siegfried Ebel, August Wilhelm Frahm, Eberhard Hackenthal, Ulrike Holzgrabe: Hagers Handbuch der Pharmazeutischen Praxis Band 8: Stoffe E-O. Springer-Verlag, 2013, ISBN 978-3-642-57994-3, S. 1032 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ B. Käsmann-Kellner: Albinismus: Weit mehr als nur blaue Augen, in: Ophthalmologe, 104 (8), 2007, S. 646–647; doi:10.1007/s00347-007-1588-8.
- ↑ R. Morell, R. A. Spritz, L. Ho, J. Pierpont, W. Guo, T. B. Friedman, J. H. Asher Jr: Apparent digenic inheritance of Waardenburg syndrome type 2 (WS2) and autosomal recessive ocular albinism (AROA)., Human Molecular Genetics, Vol. 6, S. 659–664.
- ↑ C. H. Meyer, D. J. Lapolice, S. F. Freedman: Foveal hypoplasia in oculocutaneous albinism demonstrated by optical coherence tomography. In: American journal of ophthalmology. Band 133, Nummer 3, März 2002, S. 409–410, PMID 11860983.
- ↑ A. G. Leventhal, D. J. Vitek, D. J. Creel: Abnormal visual pathways in normally pigmented cats that are heterozygous for albinism. In: Science. Band 229, Nummer 4720, September 1985, S. 1395–1397, PMID 3929383.
- ↑ Tyrosinase. In: Online Mendelian Inheritance in Man. (englisch)