Alkine

Alkine (Acetylene) sind organische Verbindungen, die mindestens eine Kohlenstoff-Kohlenstoff-Dreifachbindung (R–C≡C–R) im Molekül besitzen. Nach den IUPAC-Regeln werden solche Verbindungen allgemein als Acetylene bezeichnet, wogegen der Begriff Alkine nur für Verbindungen der Summenformel CnH2n-2 verwendet wird, also für acyclische (offenkettige) Kohlenwasserstoffe mit exakt einer Dreifachbindung. Entgegen der strengen IUPAC-Definition wird der Begriff Alkine jedoch auch für Verbindungen mit C≡C-Dreifachbindungen im Allgemeinen verwendet. In diesem weiteren Sinne fallen darunter z. B. auch Verbindungen mit zwei (Diine) oder mehr C≡C-Dreifachbindungen (Polyine) sowie Verbindungen mit einer C≡C-Dreifachbindung in einem Ring (Cycloalkine).
Alkine sind unpolare Verbindungen und ähneln mit ihren Dreifachbindungen sowohl in den physikalischen Eigenschaften als auch in der Reaktivität den Alkenen mit Doppelbindungen. Durch ihre Dreifachbindungen weisen Alkine aber eine besonders hohe Energiedichte auf und neigen unter bestimmten Bedingungen zur Polymerisation oder Explosion. Diese Tendenz nimmt umso mehr zu, je mehr Dreifachbindungen vorliegen. Man unterscheidet zwischen terminalen Alkinen, bei denen an mindestens einem Kohlenstoff-Atom der Dreifachbindung ein Wasserstoffatom anschließt, und internen Alkinen, bei denen auf beiden Seiten Kohlenstoffatome anschließen. Die Struktureinheit einer terminalen Dreifachbindung mit einer zusätzlichen Methylengruppe wird als Propargylgruppe bezeichnet. Eine wichtige Eigenschaft, die terminale Alkine von Alkanen und Alkenen unterscheidet, ist ihre vergleichsweise hohe Acidität.
Diverse Alkine kommen natürlich vor, in Pflanzen, Tieren und anderen Lebewesen und auch im Weltraum. Viele Alkine werden wegen der charakteristischen Reaktivität in der organischen Synthese eingesetzt. Das industriell wichtigste Alkin und gleichzeitig auch das einfachste ist das Ethin (Trivialname Acetylen), das im 20. Jahrhundert zeitweise eine der wichtigsten industriellen Grundchemikalien war. Obwohl seine Bedeutung seitdem abgenommen hat, wird es nach wie vor in industriellen Prozessen eingesetzt.
Geschichte
[Bearbeiten | Quelltext bearbeiten]
Die erste entdeckte natürlich vorkommende Alkinverbindung, Dehydromatricariaester, wurde 1826 aus einer Artemisia-Art isoliert, jedoch wurde die Struktur zu diesem Zeitpunkt nicht aufgeklärt.[1] 1836 versuchte Edmund Davy, der Cousin von Humphry Davy, durch Umsetzung von Weinstein mit Kohlenstoff größere Mengen Kalium zu gewinnen und stellte dabei zufällig Acetylen her. Erst etwa 20 Jahre später entwickelte Marcelin Berthelot weitere Herstellungsmethoden (insbesondere durch thermische Zersetzung von Ethylen, Ethanol und anderen organischen Verbindungen, sowie die Herstellung aus den Elementen im Lichtbogen) und gab der Verbindung den Namen Acetylen.[2][3] Im Jahr 1862 gelang dem deutschen Chemiker und Arzt Friedrich Wöhler die Darstellung von Acetylen durch die Reaktion von Wasser mit Calciumcarbid.[1] Der erste Naturstoff mit C≡C-Dreifachbindung, dessen Struktur aufgeklärt wurde, war die im Jahr 1892 isolierte Taririnsäure.[4] 1895 entdeckte Henry Le Chatelier, dass Acetylen mit Sauerstoff mit einer sehr heißen Flamme verbrennt. Damit war die Grundlage des Acetylen-Schweißens und -Schneidens gelegt.[5] In der zweiten Hälfte des 19. Jahrhunderts waren schon verschiedene Reaktionen der Alkine bekannt, darunter die Isomerisierung interner Alkine zu terminalen Isomeren, also Vorläufer der Alkin-Zipper-Reaktion,[6] und die 1869 entdeckte Glaser-Kupplung terminaler Alkine.[7]

Schon vor seiner chemisch-industriellen Nutzung wurde Acetylen für Brenner und Beleuchtungen (z. B. Autoscheinwerfer, siehe Karbidlampe) genutzt. In den 1920er und 1930er Jahren wurden Prozesse entwickelt, um aus der Verbindung verschiedene Produkte herzustellen, darunter Acetaldehyd, Trichlorethylen und Neopren. Ein besonders wichtiger Akteur war Walther Reppe, Laborchef bei I.G. Farben. Er entwickelte sicherere Prozesse zur Verarbeitung von Acetylen durch Verdünnung mit Inertgas und Herstellungsprozesse unter anderem für Propargylalkohol und Butindiol (durch Ethinylierung von Formaldehyd mit Kupferacetylid), die Folgeprodukte des Butindiols (wie Butadien und Adiponitril), sowie Cyclooctatetraen. Im Zweiten Weltkrieg wurde die Verbindung in Deutschland als Ersatz für petrochemische Materialien in der chemischen Industrie verwendet.[8] Dies ebnete dem Acetylen den Weg zu seiner großen industriellen Bedeutung Mitte des 20. Jahrhunderts. Zeitweise war es eine der wichtigsten Grundchemikalien und wurde zur Herstellung einer großen Zahl weiterer Verbindungen verwendet, darunter Acrylsäure (sowie abgeleitete Ester und Polymere), Vinylchlorid (und PVC), Vinylacetat (sowie Polyvinylacetat und andere Vinylester), Propargylalkohol und Butindiol (sowie Folgeprodukte, darunter Butan-1,4-diol, Butendiol und Tetrahydrofuran). Zum Höhepunkt der Verwendung in den 1960er und 1970er Jahren wurden in Deutschland etwa 350.000 Tonnen und in den USA 480.000 Tonnen per annum produziert. Seitdem haben die Bedeutung und die Verwendungsmenge deutlich abgenommen, da es für viele Produkte billigere Produktionsmethoden gibt, insbesondere ausgehend von Ethylen aus der Erdölverarbeitung.[9]
Etwa seit Mitte des 20. Jahrhunderts wird die C≡C-Dreifachbindung als Baustein für pharmazeutische Wirkstoffe verwendet[10] und ist ein Teil diverser Pharmazeutika, die heute verwendet werden. Zwei Nobelpreise für Chemie wurden für Forschungsthemen vergeben, bei denen Reaktionen von Alkinen einen maßgeblichen Anteil ausmachen: Durch direkte katalytische Polymerisation von Acetylen-Gas mit Titan(IV)-butanolat und Triethylaluminium als Katalysator können Polyacetylen-Folien hergestellt werden, was Ende der 1960er Jahre von Hideki Shirakawa entdeckt wurde.[11][12] Polyacetylen weist Doppel- und keine Dreifachbindungen auf und ist demnach selbst kein Alkin. Durch Dotierung (mit Chlor, Brom, Iod, Bortrifluorid, Arsen(V)-fluorid, Lithiumnaphthalid, Natrium oder Kalium) wird daraus ein Material mit hervorragender elektrischer Leitfähigkeit.[13][14][15] Die Möglichkeit der Dotierung wurde 1976 entdeckt, wobei Brom verwendet wurde.[12] Für die Entdeckung und Erforschung leitfähiger Polymere wurde 2000 der Chemie-Nobelpreis verliehen, unter anderem an Shirakawa.[16] Der Chemie-Nobelpreis 2022 wurde unter anderem für die Erfindung bzw. Erforschung der Click-Chemie verliehen, deren prototypische Reaktion die Cycloaddition zwischen einem Azid und einem terminalen Alkin ist.[17]
Vertreter und Eigenschaften
[Bearbeiten | Quelltext bearbeiten]Die Dreifachbindung der Alkine besteht aus einer σ-Bindung und zwei orthogonalen π-Bindungen. Durch die diffuse Gestalt der an den π-Bindungen beteiligten Orbitale liegt aber eine zylindrische Verteilung der Elektronen vor. Die elektronische Struktur der C≡C-Dreifachbindung hängt direkt mit der Geometrie der Dreifachbindung zusammen, in der die C-Atome der Dreifachbindung und die beiden direkt mit diesen C-Atomen verknüpften Atome in einer Linie (linear) ausgerichtet sind. Der Kohlenstoff-Kohlenstoff-Abstand einer Dreifachbindung liegt bei 120,3 pm und ist damit kürzer als der Abstand einer C=C-Doppelbindung (133 pm). Mit etwa 958 kJ/mol (gegenüber 724 kJ/mol oder 377 kJ/mol in Doppel- und Einfachbindungen) ist die Dreifachbindung auch besonders stabil, allerdings sind Alkine hochenergetische Verbindungen, die unter bestimmten Bedingungen zu Polymerisation oder Explosion neigen. Interne Alkine sind durch Hyperkonjugation energetisch etwas stabiler als terminale.[18]
Durch den höheren Anteil an s-Orbitalen in den sp-Hybridorbitalen (50 %) ist die Aufenthaltswahrscheinlichkeit der Elektronen der C–H-Bindung in der Nähe des Kohlenstoff-Kerns größer als bei Alkenen (sp2) und Alkanen (sp3), worauf die CH-Acidität endständiger (terminaler) Alkine beruht, da die negative Ladung so stabilisiert wird. Mit starken Basen können terminale Alkine deprotoniert werden und das Wasserstoffatom durch ein Metallatom ersetzt werden.[18][19][20] Beispielsweise ist der pKS-Wert der Protonen im Acetylen mit etwa 25 gegenüber 44 im Ethylen und 50 im Ethan deutlich niedriger.[18]
Lineare Alkine
[Bearbeiten | Quelltext bearbeiten]Lineare Alkine mit einer Dreifachbindung bilden eine homologe Reihe. Durch verschiedene mögliche Positionen der Dreifachbindung existieren ab vier Kohlenstoffatomen mehrere Positionsisomere. Einige Eigenschaften der Alkine bis acht Kohlenstoffatome sind in der folgenden Tabelle zusammengefasst. Ethin, Propin und 1-Butin sind bei Raumtemperatur gasförmig und 2-Butin siedet nur knapp über Raumtemperatur. Die Alkine mit fünf (Pentine) bis zehn (Decine) Kohlenstoffatomen sind alle bei Raumtemperatur flüssig. Bei gleicher Kettenlänge haben die meisten terminalen Alkine geringere Schmelz- und Siedepunkte als die internen.
| Gruppe | Summenformel | Molmasse | Isomer | Strukturformel | CAS-Nummer | Schmelzpunkt | Siedepunkt |
|---|---|---|---|---|---|---|---|
| C2H2 | 26,04 g/mol | Ethin |  |
74-86-2 | −84 °C (Sublimation)[18] | ||
| C3H4 | 40,06 g/mol | Propin | 
|
74-99-7 | −102,7 °C[21] | −23,2 °C[21] | |
| Butine | C4H6 | 54,09 g/mol | 1-Butin | 107-00-6 | −125,73 °C[22] | 8,07 °C[22] | |
| 2-Butin | 
|
503-17-3 | −32 °C[23] | 27 °C[23] | |||
| Pentine | C5H8 | 68,12 g/mol | 1-Pentin | 627-19-0 | −106 °C[24] | 39–41 °C[24] | |
| 2-Pentin | 
|
627-21-4 | −109 °C[25] | 55–57 °C[25] | |||
| Hexine | C6H10 | 82,14 g/mol | 1-Hexin | 693-02-7 | −132 °C[26] | 71 °C[26] | |
| 2-Hexin | 
|
764-35-2 | −89,6 °C[26] | 84,5 °C[26] | |||
| 3-Hexin | 928-49-4 | −103 °C[26] | 81 °C[26] | ||||
| Heptine | C7H12 | 96,17 g/mol | 1-Heptin | 628-71-7 | −81 °C[27] | 100 °C[27] | |
| 2-Heptin | 1119-65-9 | ? | 113,6 °C[28] | ||||
| 3-Heptin | 2586-89-2 | −130,5 °C[28] | 109,2 °C[28] | ||||
| Octine | C8H14 | 110,2 g/mol | 1-Octin | 629-05-0 | −79,3 °C[29] | 126,3 °C[29] | |
| 2-Octin | 2809-67-8 | −61,6 °C[29] | 137,6 °C[29] | ||||
| 3-Octin | 15232-76-5 | −103,9 °C[29] | 133,1 °C[29] | ||||
| 4-Octin | 1942-45-6 | −103 °C[29] | 131,6 °C[29] | ||||
Diine und Polyine
[Bearbeiten | Quelltext bearbeiten]
Viele Polyine mit unterschiedlichen Kettenlängen und Substituenten wurden untersucht. Schon Butadiin (Diacetylen) mit zwei Dreifachbindungen ist sehr reaktiv und Hexatriin (Triacetylen) ist extrem instabil,[30] kommt allerdings natürlich als Metabolit des Pilzes Fomes annosus vor.[31] Eine in-silico-Studie von 1,3-Butadiin und 1,3-Pentadiin zeigt, dass im Gegensatz zu den analogen Verbindungen mit konjugierten Doppelbindungen keine nennenswerte Resonanzstabilisierung vorliegt.[32] Je länger Polyine sind, also je mehr Dreifachbindungen sie aufweisen, desto instabiler und reaktiver sind sie, da sie leicht exotherm polymerisieren und sich insbesondere als Feststoffe explosiv zersetzen können, auch in Abwesenheit von Luft. Terminal substituierte Polyine sind stabiler, wobei die Stabilität mit der Größe der Substituenten zunimmt, da größere Gruppen eine Annäherung der Ketten effektiv verhindern können.[30] Während Hexatriin schon unter Raumtemperatur explodiert, kann Diphenylhexatriin erhitzt und geschmolzen werden. tert-Butylgruppen haben ebenfalls einen deutlich stabilisierenden Effekt, wodurch schon in den 1950er-Jahren Di-tert-butyltetradecaheptain hergestellt werden konnte.[30][33][34][35] Erst 2010 wurde ein Polyin mit 22 Dreifachbindungen und Tris(3,5-di-tert-butylphenyl)methyl-Gruppen als endständige Substituenten hergestellt. Im Jahr 2020 wurde ein noch längeres Polyin mit 24 Dreifachbindungen präsentiert. Ein sehr langkettiges Polyin ist das Carbin.[33][36]
Cycloalkine
[Bearbeiten | Quelltext bearbeiten]
Cycloalkine mit kleineren Ringgrößen weisen eine erhebliche Ringspannung auf. Cyclooctin wurde in den 1950er-Jahren zum ersten Mal synthetisiert und ist das kleinste Cycloalkin, das unter normalen Bedingungen isoliert werden kann (Siedepunkt 157,5–158 °C), obwohl es auch sehr reaktiv ist.[37][38][39][40] Bei dieser Verbindung beträgt der Winkel der Alkinsubstituenten etwa 160° und die Ringspannung beträgt nach unterschiedlichen Angaben 9–19 kcal/mol.[38][41][42] Cyclopentin, Cyclohexin und Cycloheptin sind als Liganden von Übergangsmetallkomplexen bekannt.[37] Die Halbwertszeit von freiem Cycloheptin beträgt etwa eine Stunde bei −78 °C, freies Cyclohexin konnte bisher nur durch Matrixisolation bei −100 °C untersucht werden und freies Cyclopentin ist lediglich als reaktives Intermediat bekannt.[43][44] Cyclobutin wurde nur theoretisch untersucht und existiert möglicherweise nur als Übergangszustand.[45][46] Das kleinste Cycloalkin ohne Ringspannung ist Cyclodecin.[47]
Spektroskopische Eigenschaften
[Bearbeiten | Quelltext bearbeiten]Im 1H-NMR sind Alkinyl-Wasserstoffatome nicht so stark entschirmt wie Alkenylwasserstoffatome, da in der rotations-symmetrischen Dreifachbindung durch das äußere Magnetfeld ein Ringstrom induziert wird, dessen Magnetfeld dem äußeren entgegengesetzt ist.[18] Daher erscheinen Wasserstoffatome terminaler Alkine bei 1,7–3,3 ppm (2–2,5 ppm).[18][19] Bei Protonen terminaler Alkine liegt oft eine Fernkopplung (4J-Kopplung) von etwa 2–4 Hz über die Dreifachbindung hinweg vor.[18][19] Im 13C-NMR erscheinen die Kohlenstoffatome, die an der Dreifachbindung eines Alkins beteiligt sind, bei etwa 65–95 ppm, also bei höherer Verschiebung als Alkane aber niedrigerer als Alkene.[18] Die Verschiebung hängt deutlich vom Winkel der Alkinsubstituenten ab: Bei Cyclooctin mit einem Winkel von etwa 160° ist die Verschiebung um 95 ppm. Bei stärker gebogenen Verbindungen mit einem Winkel von 140–150° kann die Verschiebung auch 100–110 ppm betragen.[48]
Auch durch Infrarotspektroskopie können Alkine nachgewiesen werden. Die C–H-Bindung weist eine Valenzschwingung bei 3260–3330 cm−1 und eine Deformationsschwingung bei etwa 640 cm−1 auf. Die C≡C-Bindung weist eine Valenzschwingung bei 2100–2260 cm−1 auf, die aber bei internen Alkinen weniger deutlich ist, wodurch Infrarotspektroskopie insbesondere zum Nachweis terminaler Alkine geeignet ist.[18]
Natürliche Vorkommen
[Bearbeiten | Quelltext bearbeiten]Alkine sind als Naturstoffe nicht sehr verbreitet, mit Stand des Jahres 2013 waren lediglich etwa 1000 Verbindungen bekannt[49], 2019 jedoch schon über 2000.[50] Allerdings sind diese Verbindungen auf sehr unterschiedliche Lebewesen verteilt und kommen sowohl in Pflanzen, Tieren und Pilzen als auch in Mikroorganismen vor.
Vorkommen in Pflanzen
[Bearbeiten | Quelltext bearbeiten]Neben der Taririnsäure sind diverse weitere Fettsäuren mit Dreifachbindungen bekannt, darunter die Crepeninsäure, die in Korbblütlern vorkommt (z. B. Crepis foetida),[51][52] und die Ximeninsäure, die in Sandelholzartigen (z. B. Santalum album) vorkommt.[53][54] In der Familie der Korbblütler kommen neben der Crepeninsäure viele weitere Alkine vor, darunter auch eine große Zahl an Verbindungen mit mehreren Dreifachbindungen.[49][55] Das Haupttoxin der Wasserschierlinge (Cicutoxin) ist ebenfalls ein Alkin.[56] Der Polyol Avocadin aus der Avocado trägt eine terminale Dreifachbindung.[57]
-
 Crepis foetida
Crepis foetida -
 Crepeninsäure
Crepeninsäure -
 Santalum album
Santalum album -
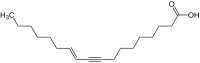 Ximeninsäure
Ximeninsäure -
 Wasserschierling
Wasserschierling -
 Cicutoxin
Cicutoxin -
 Avocado
Avocado -
 Avocadin
Avocadin
.jpg)

.jpg)



Vorkommen in Tieren
[Bearbeiten | Quelltext bearbeiten]

1971 wurden die ersten Vertreter der Histrionicotoxine aus dem Harlekin-Baumsteiger, einem Pfeilgiftfrosch, isoliert. Die meisten Vertreter dieser Gruppe weisen eine Dreifachbindung auf, manche, wie das Histrionicotoxin selbst, sogar zwei.[58]
Eine große Anzahl an Alkinen wurde aus Schwämmen isoliert. Beispielsweise wurden mehr als zehn langkettige, lineare, stark ungesättigte Verbindungen mit jeweils vier Dreifachbindungen aus einer Art der Gattung Petrosia isoliert[59] und auch weitere Polyine wurden in dieser Gattung gefunden.[60][61] Callyspongia[62][63] und Stelletta[63] sind weitere Gattungen von Schwämmen, aus denen Alkine isoliert wurden. Auch in Steinkorallen der Gattung Montipora kommen Alkine vor.[59][62]
Vorkommen in Pilzen
[Bearbeiten | Quelltext bearbeiten]In Pilzen kommen viele Alkin-Metaboliten vor[49], darunter Carbonsäuren wie das Mycomycin[64], Epoxide wie Biformin (aus Trichaptum biforme)[49][S 1], Alkinylphenole wie das Siccain[49][S 2] und einfache Kohlenwasserstoffe wie das Hexatriin.[31]
-
 Der Pilz Fomes annosus bildet flüchtiges Hexatriin
Der Pilz Fomes annosus bildet flüchtiges Hexatriin -
 Trichaptum biforme
Trichaptum biforme


-
 Mycomycin
Mycomycin -
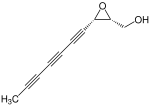 Biformin
Biformin -
 Siccain
Siccain



Vorkommen in Mikroorganismen
[Bearbeiten | Quelltext bearbeiten]In vielen marinen Cyanobakterien kommen Peptide vor, die Komponenten mit terminaler Dreifachbindung enthalten, beispielsweise Onchidin[S 3] und die Dragonamide.[65]
Eine große Gruppe von biologisch aktiven Alkinen bilden die Endiin-Antibiotika, die seit den späten 1980er-Jahren erforscht werden. Naturstoffe, die als Strukturmotiv die Endiin-Einheit besitzen, wirken häufig cytotoxisch gegenüber menschlichen Tumorzelllinien und stellen somit potentielle Chemotherapeutika dar. Der Wirkmechanismus beruht auf einer Bergman-Cyclisierung (siehe unten).[66][67] Etwa 40 natürliche Endiin-Verbindungen sind bekannt.[66] Beispiele hierfür sind das von Streptomyces carcinostaticus sezernierte Neocarzinostatin[68], die Calicheamicine aus Micromonospora echinospora[69], die Esperamicine aus Actinomadura verrucospora[70] und das Dynemicin aus Micromonospora chersina.[71]
Vorkommen im Weltall
[Bearbeiten | Quelltext bearbeiten]
In den protoplanetaren Nebeln CRL 618 und CRL 2688 wurden Acetylen, Butadiin (Diacetylen) und Hexatriin (Triacetylen) nachgewiesen.[72] In der Atmosphäre des Saturnmonds Titan wurden ebenfalls Acetylen und Diacetylen sowie Propin nachgewiesen.[73][74] Auch im interstellaren Raum wurden diverse Alkine nachgewiesen, darunter Ethin, Propin, Diacetylen, 1,3-Pentadiin, Hexatriin sowie Cyanoacetylen und diverse Cyanopolyine mit zwei bis fünf konjugierten Dreifachbindungen.[75]
-
 Diacetylen
Diacetylen -
 Pentadiin
Pentadiin -
 Cyanotriacetylen, ein Cyanopolyin
Cyanotriacetylen, ein Cyanopolyin
Nomenklatur
[Bearbeiten | Quelltext bearbeiten]Gemäß den Regeln der IUPAC werden Verbindungen mit Dreifachbindungen im Allgemeinen als Acetylene bezeichnet[76], während der Begriff Alkine nur für eine eng umgrenzte Gruppe von Verbindungen gilt, die acyclisch (offenkettig) sind, exakt eine C≡C-Dreifachbindung aufweisen und demnach die Summenformel CnH2n–2 besitzen.[77][78] Acyclische verzweigte oder unverzweigte Kohlenwasserstoffe mit mehr als einer Dreifachbindung werden als Alkadiine, Alkatriine usw. bezeichnet.[77] Entgegen den IUPAC-Regeln wird jedoch auch der Begriff Alkine im weiteren beziehungsweise allgemeinen Sinn gebraucht.[79][80]
Die Benennung der Alkine nach den IUPAC-Regeln orientiert sich an den Namen für die Alkane. Als Stammnamen für das Alkin wählt man den Wortstamm des Alkans mit gleicher Anzahl an Kohlenstoffatomen und ersetzt die Endsilbe -an durch -in. Bei verzweigten Alkinen gibt die längste mögliche Kohlenstoffkette mit der Dreifachbindung den Stammnamen. Die Lage der Dreifachbindung wird mit einer vorgestellten Zahl beschrieben, sodass diese möglichst klein ist z. B. 1-Butin (statt 3-Butin) für H–C≡C–CH2–CH3 und 2-Pentin (statt 3-Pentin) für H3C–C≡C–CH2–CH3. Enthält die Kohlenstoffkette mehrere Dreifachbindungen, so fügt man im Namen vor der Silbe in die Silbe di, tri, tetra usw. ein. So erhält ein Alkin mit fünf (griechisch penta) C-Atomen und zwei Dreifachbindungen nach dem 1. und 4. C-Atom den IUPAC-Namen 1,4-Pentadiin (H–C≡C–CH2–C≡C–H). Das strukturisomere 1,3-Pentadiin hat demnach folgende Struktur: H–C≡C–C≡C–CH3. Gibt es für eine Verbindung mit einer Doppel- und einer Dreifachbindung zwei unterschiedliche Benennungsmöglichkeiten mit dem gleichen Lokantensatz (Zahlenkombination), z. B. bei H–C≡C–CH2–CH=CH2 dann erhält die Doppelbindung die kleinere Ziffer, sodass der korrekte Name Pent-1-en-4-in[S 4] (statt Pent-4-en-1-in) ist.[18]
Herstellung
[Bearbeiten | Quelltext bearbeiten]Alkine können analog zu Alkenen durch Eliminierungsreaktionen gewonnen werden.[18] Aus terminalen Alkinen können außerdem komplexere Alkine aufgebaut werden, beispielsweise durch Additions- oder Substitutionsreaktionen oder durch eine Sonogashira-Kupplung, siehe Abschnitt Reaktionen terminaler Alkine.
Eliminierung
[Bearbeiten | Quelltext bearbeiten]Alkine erhält man durch Dehydrohalogenierung von Dihalogenalkanen oder Alkenylhalogeniden mit verschiedenen Basen.[81][20] Beispielsweise können diese durch Eliminierung von Bromwasserstoff aus einem Alkenylbromid mit geeignetem Substitutionsmuster gewonnen werden. Die beiden Atome müssen antiperiplanar zueinander stehen, das heißt jeweils eines an jedem Atom der Doppelbindung, in (E)-Stellung. Die zweifache Eliminierung aus einem Dihalogenalkan (z. B. 1,2-Dibromalkan) mit einem Alkenylhalogenid als Zwischenstufe ist ebenfalls möglich. So kann durch Umsetzung von 1,2-Dibrompropan mit Lithiumdiisopropylamid Propin gewonnen werden.[18][19] Andere geeignete Basen sind Natriumamid und Kalium-tert-Butanolat.[18] Während der erste Eliminierungsschritt auch mit weniger starken Basen funktioniert, sind für den zweiten Schritt drastischere Reaktionsbedingungen nötig.[20] Durch Halogenierung und anschließende zweifache Dehydrohalogenierung kann ein Alken in ein Alkin überführt werden.[18] Beispielsweise kann Ölsäure in Dichlorstearinsäure oder Dibromstearinsäure überführt werden, deren zweifache Dehydrohalogenierung Stearolsäure ergibt.[82][83][84]

oder durch Bromierung Dibromstearinsäure (unten) erhalten werden.
Aus beiden Halogensäuren kann durch zweifache Dehydrohalogenierung
Stearolinsäure erhalten werden
Alkinsynthese nach Bourguel
[Bearbeiten | Quelltext bearbeiten]Bei der Alkinsynthese nach Bourguel wird zunächst 2,3-Dibrom-1-propen mit einer Grignard-Verbindung umgesetzt. Anschließende Eliminierung mit Natriumamid führt zum Alkin.[85]
Herstellung aus Carbonylen
[Bearbeiten | Quelltext bearbeiten]
Es sind verschiedene Methoden bekannt, um Carbonyle, also Aldehyde und Ketone in Alkine umzuwandeln, wobei jeweils ein zusätzliches Kohlenstoffatom eingeführt wird. Bei der Corey-Fuchs-Reaktion wird zunächst ein Aldehyd mittels Triphenylphosphin und Tetrabrommethan zu einem gem-Dibromolefin umgesetzt. Eliminierung mittels Butyllithium erzeugt ein Lithiumacetylid. Dieses kann durch Hydrolyse in ein terminales Alkin überführt werden oder mittels Kohlenstoffdioxid in eine Carbonsäure.[86][87] Eine Umsetzung mit anderen Elektrophilen wie Alkylhalogeniden, Aldehyden und Epoxiden ist aber auch möglich.[86][88] Durch Verwendung einer geeigneten Base (Natriumhexamethyldisilazid) kann das Dibromolefin auch in ein Bromalkin überführt werden.[88] Eine Eintopfreaktion ist unter Verwendung von Dibrommethyltriphenylphosphoniumbromid möglich.[89] Der Vorteil dieser Reaktion ist die einfache Anpassbarkeit durch Verwendung eines bestimmten Elektrophils. Nachteil ist die Notwendigkeit zum Einsatz starker Basen sowie die schwierige Aufarbeitung durch Nebenprodukte wie Triphenylphosphinoxid.[86][88] Eine ähnliche Reaktion verwendet Hexamethylphosphorsäuretrisamid und Bromtrichlormethan, aus denen ein gem-Dichloralken erzeugt wird.[88]
Bei der Colvin-Umlagerung wird Trimethylsilyldiazomethan oder Dimethyldiazomethylphosphonat (DAMP) als Kohlenstoffquelle und Lithiumdiisopropylamid oder Butyllithium als Base eingesetzt.[86][90][91] Vorteil dieser Reaktion ist die Verwendung von Trimethylsilyldiazomethan, welches ein sehr einfaches und käuflich erhältliches Reagenz ist. Nachteil sind einerseits die Notwendigkeit zum Einsatz starker Basen und tiefer Temperaturen, andererseits die Nukleophilie des Reagenzes durch die es inkompatibel mit diversen funktionellen Gruppen ist.[86]
Bei der Seyferth-Gilbert-Homologisierung werden DAMP und Kalium-tert-Butanolat verwendet, bei der Weiterentwicklung nach Ohira und Bestmann das sogenannte Ohira-Bestmann-Reagenz, aus dem DAMP durch Umsetzung mit Kaliumcarbonat in Methanol in situ gewonnen werden kann. Die Reaktion gelingt an Luft bei Raumtemperatur und ohne starke Basen. Vorteile sind die milden Bedingungen und einfache Aufarbeitung. Nachteil ist dabei, dass die Reaktion weder mit Ketonen noch mit α,β-ungesättigten Aldehyden funktioniert.[86] Das Ohira-Bestmann-Reagenz ist vergleichsweise teuer. Ein alternatives Reagenz, das statt einer Methylgruppe eine Phenylgruppe trägt, kann leicht durch Umsetzung von 2-Bromacetophenon mit Trimethylphosphit und Mesylazid hergestellt werden.[92]
Viele weitere Phosphorreagenzien zur Erzeugung von Alkinen aus Carbonylen sind ebenfalls bekannt.[88]
Herstellung von Polyinen
[Bearbeiten | Quelltext bearbeiten]Polyine werden oft durch Kombinationen von Eliminierungs- und Kupplungsreaktionen (beispielsweise Glaser-Eglinton-Hay-Reaktionen) hergestellt.[30][33]
Polyine können außerdem durch Laserablation aus Grafit hergestellt werden, wobei in der Regel Gemische anfallen. Beispielsweise entstehen unter einer Argon-Propan-Atmosphäre Hexatriin, Octatetrain und weitere Polyine bis C14.[93] In wässriger Phase entstehen beispielsweise Hexatriin, Octatetrain und Decapentain.[94] Durch Laserablation aus suspendierten Grafit-Partikeln werden je nach Lösungsmittel andere Gemische enthalten. In Hexan werden vorwiegend Polyine mit C8 bis C14 erhalten. In Benzol oder Toluol vor allem solche mit C10 bis C16.[95] Durch einen Lichtbogen zwischen Grafit-Elektroden in Methanol oder Ethanol werden ebenfalls Polyine, insbesondere Octatetrain, erhalten.[96]
Herstellung von Cycloalkinen
[Bearbeiten | Quelltext bearbeiten]Cycloalkine mit größeren Ringgrößen können durch diverse Ringschlussreaktionen erzeugt werden, kleinere hingegen fast nur durch Eliminierungsreaktionen.[48] Beispiele für Ringschlussreaktionen, mit denen Cycloalkine erzeugt werden können, sind die Alkinmetathese[97] und die Glaser-Kupplung.[98] Cyclische Diine können erzeugt werden aus einer Alkylkomponente, die zwei terminale Halogenatome trägt und einer Komponente, die zwei terminale Dreifachbindungen trägt. Durch Umsetzung mit Butyllithium wird ein Acetylid erzeugt, das mit dem Halogenid eine Substitutionsreaktion eingeht.[99] Ein Beispiel für eine Synthese durch Eliminierung ist die Herstellung von Cyclooctin durch Bromierung und zweifache Dehydrobromierung von Cycloocten.[40]

Reaktionen der Alkine
[Bearbeiten | Quelltext bearbeiten]Alkine gehen viele charakteristische Reaktionen ein sowie diverse Reaktionen, die denen von Alkenen analog sind. Da Alkine in vielfältigen Reaktionen umgesetzt werden können und Ausgangsprodukte vieler Namensreaktionen sind, spielen sie eine wichtige Rolle in der organischen Synthese und wurden zum Beispiel für die Synthesen verschiedener Naturstoffe eingesetzt.
Hydrierung
[Bearbeiten | Quelltext bearbeiten]Alkine können analog zu Alkenen durch Wasserstoff hydriert werden. Mit den für Alken-Hydrierungen üblichen Katalysatoren (Palladium oder Platin auf Kohle) werden dabei Alkane erhalten.[18][20]
Durch geeignete Reaktionsbedingungen können Alkine selektiv sowohl zu (Z)-Alkenen als auch zu (E)-Alkenen hydriert werden. Mit einem vergifteten Katalysator, dem Lindlar-Katalysator, reagiert das Alkin nur zum Alken. Dabei entstehen ausschließlich (Z)-Alkene (cis), da sich die Wasserstoffatome von der gleichen Seite an das Alkin nähern und reagieren. Geeignete Katalysatorsysteme sind beispielsweise Palladium auf Calciumcarbonat mit Blei(II)-acetat als Katalysatorgift oder Palladium auf Bariumsulfat mit Chinolin als Katalysatorgift. Dabei dient jeweils das Katalysatorgift der Herabsetzung der katalytischen Aktivität. Die dabei verwendeten Unterlagematerialien sind ebenfalls wichtig. Da sie Produkte schneller freisetzen als die normalerweise verwendete Kohle, tritt weniger Überreaktion auf.[19]
Die Hydrierung zu einem (E)-Alken ist über eine radikalische Reaktion mit elementarem Natrium in flüssigem Ammoniak möglich[19], mit Lithium in Methyl-, Ethyl- oder Propylamin[100] oder mit Natrium und tert-Butanol in Hexamethylphosphorsäuretrisamid.[101] Eine Alternative ist die Reduktion mit Lithiumaluminiumhydrid oder Red-Al, die jedoch nur funktioniert, wenn sich in der Nähe der Dreifachbindung ein Sauerstoffatom (z. B. eine Hydroxylgruppe) befindet, das das Aluminium koordinieren kann.[19]

Elektrophile Addition
[Bearbeiten | Quelltext bearbeiten]Analog zu den Doppelbindungen in Alkenen können diverse Elektrophile an die Dreifachbindungen in Alkinen addiert werden. Beispielsweise können Alkine bromiert werden, wobei in der Regel ebenfalls ein Bromonium-Ion als Zwischenstufe entsteht.[19]
Hydrohalogenierung
[Bearbeiten | Quelltext bearbeiten]Alkine können mit Bromwasserstoff oder Iodwasserstoff hydrohalogeniert werden. Allerdings ist die Reaktion mit Bromwasserstoff nicht selektiv, da eine radikalische Nebenreaktion auftritt, die nur durch Ausschluss von Licht und Sauerstoff sowie Zusatz eines Radikalfängers verhindert werden kann. Chlorwasserstoff ist gegenüber den meisten Alkinen unreaktiv. Zusatz von Aluminiumoxid oder Silica zur Reaktion ermöglicht aber die selektive Hydrohalogenierung mit Chlorwasserstoff und Bromwasserstoff.[102] Solche Hydrohalogenierungen sind im Allgemeinen aber nicht stereoselektiv, da (E)- und (Z)-Isomer im Gleichgewicht vorliegen, wobei zuerst das kinetisch vorteilhafte (Z)-Isomer entsteht, das aber überwiegend zum thermodynamisch stabileren (E)-Isomer isomerisiert.[102][103] Chlorwasserstoff oder Bromwasserstoff kann für die Reaktion in situ gewonnen werden aus Vorläufermolekülen wie Thionylchlorid, Oxalylchlorid, Oxalylbromid, Phosphortribromid oder Acetylbromid.[103]
Hydratisierung
[Bearbeiten | Quelltext bearbeiten]Die Oxymercurierung von Alkinen ergibt Enole, die sich zügig zu Ketonen umlagern. Aus terminalen Alkinen können so Methylketone gewonnen werden.[19][20][103] Aus internen Alkinen entstehen im Regelfall zwei regioisomere Ketone.[103] Addition von Essigsäure an Alkine ergibt Enolacetate, die schnell hydrolysieren und sich dann ebenfalls zu Ketonen umlagern.[104] Die Hydroborierung von Alkinen erfordert geeignete Boran-Reagenzien, um Polymerisationen als Nebenreaktionen zu vermeiden. Geeignet ist zum Beispiel Catecholboran. Das entstehende Alkenylboran kann je nach Reaktionsbedingungen in weitere nützliche Verbindungen überführt werden: Mit Wasser in eine Alkenylboronsäure, mit Brom und einer Base in ein Alkenylbromid, mit Wasserstoffperoxid über ein Enol in ein Keton und mit Essigsäure in ein (Z)-Alken.[103]
Alkin-Zipper-Reaktion
[Bearbeiten | Quelltext bearbeiten]Die Alkin-Zipper-Reaktion ist eine Isomerisierung, durch die mittels Kalium-(3-aminopropylamid) (hergestellt aus 1,3-Diaminopropan und Kaliumhydrid) interne Alkine zu terminalen Alkinen umgesetzt werden können, die wichtige synthetische Intermediate sind.[6][105] Eine ähnliche Isomerisierung mit verschiedenen starken Basen wurde erstmals Ende des 19. Jahrhunderts beschrieben, allerdings ermöglichte diese nur die Wanderung einer Dreifachbindung über ein bis zwei Bindungen bei sehr hohen Temperaturen und langen Reaktionszeiten, während die moderne Reaktion auch eine Wanderung über zehn Bindungen bei 0 °C in sehr kurzen Reaktionszeiten (z. T. innerhalb von Sekunden) ermöglicht.[6][106] Die Isomerisierung zu einem terminalen Alkin ist thermodynamisch ungünstig, allerdings fällt durch Deprotonierung des terminalen Alkins ein Acetylid-Salz aus, was die Triebkraft der Reaktion darstellt.[105][106] Mit dem analogen Natriumreagenz ist die Reaktion ebenfalls möglich, wodurch die Verwendung von gefährlichem Kaliumhydrid vermieden werden kann, allerdings funktioniert diese Reaktion erst bei höheren Temperaturen von 50-60 °C und erfordert längere Reaktionszeiten.[6][105] Die Methode wurde in diversen Naturstoffsynthesen verwendet, beispielsweise von Muscon[107] und von (9Z, 25S, 26R, 43Z)-25,26-Epoxy-9,43-henpentacontadien, einem Pheromon der Kakerlake Nauphoeta cinerea.[108] Eine Synthese von Avocadin ging von einer Propargylverbindung und 1-Iodundecan aus; durch anschließende Isomerisierung wurde die terminale Dreifachbindung aufgebaut.[109] Bei einer Synthese verschiedener Mycolsäuren wurde das kommerziell erhältliche 3-Heptin-1-ol als Edukt in das terminale Isomer 6-Heptin-1-ol überführt.[110]

Cyclisierungen
[Bearbeiten | Quelltext bearbeiten]Anhand der Baldwin-Regeln[111] und darauf aufbauender weiterentwickelter Regeln lässt sich vorhersagen, ob eine Cyclisierungsreaktion geometrisch vorteilhaft ist. Dies hängt maßgeblich davon ab, welchen Hybrisierungszustand das beim Ringschluss angegriffene Atom hat (bei Dreifachbindungen sp, was als dig, für digonale Geometrie bezeichnet wird), welche Größe der gebildete Ring hat und ob eine dabei gebrochene Bindung Teil des Rings wird (bei Dreifachbindungen ist dies eine Doppelbindung, die durch Bruch der zweiten π-Bindung entsteht).[112][113][114] Ein Beispiel für eine Cyclisierungsreaktion mit derart vorhersehbarer vorteilhafter Geometrie ist der durch Gold(I) katalysierte Aufbau des Pyridinrings mit exocyclischer Doppelbindung bei der Totalsynthese von Conolidin.[115]
Durch eine stufenweise Cyclotrimerisierung kann aus drei Alkinmolekülen ein aromatischer Ring aufgebaut werden.[116] Dabei handelt es sich um eine konvergente, atomeffiziente Methode, um insbesondere hochsubstituierte Aromaten herzustellen, allerdings ist die Regioselektivität oft schwierig zu kontrollieren, insbesondere, wenn zwei oder sogar drei unterschiedliche Alkinkomponenten verwendet werden.[117] Als Katalysator für Cyclotrimerisierungen wird insbesondere Ruthenium verwendet.[116][117]
Durch Umsetzung von Alkinen mit Nitriloxiden können Isoxazole gewonnen werden.[19] Auch andere 1,3-dipolare Cycloadditionen von Alkinen spielen eine Rolle, zum Beispiel die Addition von Aziden und die Ozonolyse, siehe unten.
Diels-Alder-Reaktionen
[Bearbeiten | Quelltext bearbeiten]Werden bei Diels-Alder-Reaktionen eine oder mehrere Doppelbindungen der Edukte durch Dreifachbindungen ersetzt, ergibt die Reaktion gespannte cyclische Allene, die sich im Allgemeinen weiter umlagern, z. B. durch Wanderungen von Wasserstoffatomen, wodurch die Ringspannung reduziert wird. Während die normale Diels-Alder-Reaktion fast immer ein konzertierter thermischer Prozess ist, sind Diels-Alder-Reaktionen mit Alkinen oft mehrstufig und es sind sowohl thermische als auch photochemische Reaktionen bekannt, wobei letztere typischerweise bei Raumtemperatur ablaufen. Im Allgemeinen entstehen als Endprodukte aromatische Ringe, bei der Umsetzung eines Diens mit einem Alkin über die Zwischenstufe eines 1,4-Cyclohexadiens.[118][119] Bei Umsetzung eines Diins mit einem Alkin entsteht ein hochreaktives Arin.[119][120] Diels-Alder-Reaktionen von Alkinen können beispielsweise zum Aufbau von polycyclischen aromatischen Verbindungen genutzt werden.[121]
Pauson-Khand-Reaktion und Dötz-Reaktion
[Bearbeiten | Quelltext bearbeiten]Die Pauson-Khand-Reaktion ermöglicht die Synthese eines Cyclopentenons aus einem Alken, einem Alkin und Kohlenmonoxid mittels Dicobaltoctacarbonyl. Die Reaktion benötigt oft hohe Temperaturen, vermutlich wegen der nötigen Abspaltung eines Kohlenmonoxid-Liganden, durch den eine Bindung des Alkens oder Alkins an das Cobalt möglich wird. Durch Zugabe eines tertiären Aminoxids (z. B. N-Methylmorpholin-N-oxid oder Trimethylaminoxid), kann ein Kohlenmonoxidligand als Kohlendioxid abgespalten werden und die Reaktion ist unter deutlich milderen Bedingungen möglich.[122]
Bei der 1975 entdeckten Dötz-Reaktion handelt es sich formal um eine [3+2+1]-Cycloaddition. Edukte sind ein α,β-ungesättigtes Fischer-Carben (mit Aryl- oder Vinylgruppe) und ein Alkin. Durch Einbau eines Kohlenmonoxid-Moleküls entsteht ein Chinon. Die Reaktion hat große Bedeutung bei der Synthese von Naturstoffen mit einer 1,4-Naphthochinon-Struktureinheit.[123]
Bergman-Cyclisierung und verwandte Reaktionen
[Bearbeiten | Quelltext bearbeiten]Eine spezielle Reaktion von Endiinen ist die Bergman-Cyclisierung, bei der sich diese Struktur zu einem Benzol-1,4-Diradikal umlagert.[124] Die Aktivierungsenergie hängt dabei stark von der konkreten Struktur ab.[125] Während die 1972 von Bergman beschriebene Reaktion von Hexa-1,5-diin-3-en hohe Temperaturen benötigt[126], können verschiedene Naturstoffe, die später entdeckt wurden und ebenfalls eine Endiin-Struktur aufweisen, eine solche Reaktion schon bei Raumtemperatur eingehen.[125] Verschiedene Faktoren spielen eine Rolle für die Aktivierungsenergie, darunter der Abstand zwischen den beiden Dreifachbindungen, Substituenten an den Dreifachbindungen und ein Abbau von Ringspannung.[67][124] Letzteres ist zum Beispiel bei der Bergman-Cyclisierung von Calicheamicin γ1 wichtig.[67]
Bei der verwandten Moore-Cyclisierung, die ebenfalls über ein Diradikal verläuft, reagiert ein Enin-Keten zu einem Chinon.[127] Eine analoge Reaktion ohne Beteiligung von Sauerstoffatomen ist die Myers-Cyclisierung, bei der ein Enin-Allen zu einem Benzol-Ring reagiert.[128]
Oxidationen
[Bearbeiten | Quelltext bearbeiten]Durch bestimmte Oxidationsmittel wie Kaliumpermanganat oder Ruthenium(IV)-oxid mit Natriumperiodat können Alkine zu Dionen oxidiert werden.[103] Eine oxidative Spaltung ist ebenfalls möglich, beispielsweise mit Chromsäure[20], durch Ozonolyse, sowie mit Ruthenium(VIII)-oxid oder Osmium(VIII)-oxid.[129] Eine weitere Methode der oxidativen Spaltung verwendet Eisen(III)-chlorid und tert-Butylhydroperoxid.[130] Im Allgemeinen entstehen bei solchen oxidativen Spaltungen Carbonsäuren, aber unter bestimmten Bedingungen können auch Carbonsäureester oder Carbonsäureamide erhalten werden.[129]
Metathesen
[Bearbeiten | Quelltext bearbeiten]
Bei der Alkinmetathese werden analog zur Olefinmetathese zwischen zwei Alkinen die Enden der Dreifachbindungen getauscht.[97] Die erste solche Reaktion, die beschrieben wurde, nutzte einen heterogenen Katalysator aus Oxiden des Wolframs auf Silica und funktionierte nur bei hohen Temperaturen über 200 °C.[97][131] Die erste Reaktion mit homogenem Katalysator wurde von Mortreux beschrieben, der p-Tolylphenylacetylen mit Molybdänhexacarbonyl und Resorcin umsetzte, wodurch Diphenylacetylen und Di-p-tolylacetylen erhalten wurden.[97][132] Die meisten Katalysatoren der Alkinmetathese sind gegenüber Alkenen unreaktiv. Der Mechanismus besteht in einer [2+2]-Cycloaddition und Cycloreversion mit einem Metallacyclobutan als Intermediat. Ein typischer Katalysator ist Tri-tert-butoxy(tert-butylmethylidin)wolfram(VI), das kommerziell erhältlich ist. Die Alkinmetathese kann auch in Form einer Ringschlussmetathese durchgeführt werden. Bei Alkenmetathesen ist eine Stereoselektivität oft schwierig zu erzielen. Mit einer Alkinmetathese und Lindlar-Hydrierung ist hingegen problemlos ein (Z)-Alken zu erhalten. Sie wurde daher für diverse Totalsynthesen eingesetzt.[97] Beispiele für Naturstoffe, die so synthetisiert wurden, sind Zibeton[133], sowie Ambrettolid und Yuzu-Lacton.[134]
Bei der Enin-Metathese werden ein Alken und ein Alkin zu einem 1,3-Dien umgesetzt. Die Reaktion ist mit der Olefin-Metathese verwandt und in vielen Fällen kommen dieselben Katalysatoren wie bei Olefinmetathesen zum Einsatz, z. B. Grubbs-Katalysatoren. Eines der wichtigsten Einsatzgebiete dieser Reaktion ist die Synthese von cyclischen Molekülen. Die Methode wurde für verschiedene Naturstoffsynthesen eingesetzt, zuerst von Stemoamid[135], außerdem zum Beispiel von Anatoxin A.[136][137]
Polymerisationen
[Bearbeiten | Quelltext bearbeiten]Acetylengas kann mit Titan(IV)-butanolat und Triethylaluminium als Katalysator zu Polyacetylen polymerisiert werden.[12] Diverse Reaktionen von Alkinen können mit geeigneten Monomeren zur Herstellung von Polymeren verwendet werden, beispielsweise die Sonogashira-Kupplung und die Glaser-Hay-Kupplung.[138] Polykupplungen von Alkinen können beispielsweise mit Kupfer(I)-chlorid oder Indium(III)-chlorid katalysiert werden.[139] Die Polyaddition von Nukleophilen an Alkine ist eine einfache Methode zur Herstellung von Vinylen-Polymeren.[138] Durch Polycyclotrimerisierung (z. B. mittels Tantal(V)-chlorid und Tetraphenylstannan oder mittels Tantal(V)-bromid) können hyperverzweigte Netze aus aromatischen Ringen aufgebaut werden.[140] Auch eine Polymerisation durch Click-Chemie ist mit geeigneten Monomeren möglich.[141][142]
Nicholas-Reaktion
[Bearbeiten | Quelltext bearbeiten]Bei der Nicholas-Reaktion wird ein Dicobalthexacarbonylkomplex eines Propargylalkohols mit Säure umgesetzt, wodurch ein Propargylkation erhalten wird. Dieses kann anschließend mit diversen Nucleophilen umgesetzt werden, darunter Alkohole, Amine, Thiole, Aluminiumalkyle, Hydridüberträger (wie Tributylzinnhydrid oder Natriumborhydrid), Silylenolether oder Allyltributylzinn.[143]
Reaktionen von Cycloalkinen
[Bearbeiten | Quelltext bearbeiten]In pericyclischen Reaktionen mit Aziden werden typischerweise terminale Alkine unter Kupferkatalyse verwendet (s. u.), es sind jedoch auch kupferfreie Varianten mit Cyclooctinen bekannt, wobei die Triebkraft der Abbau von Ringspannung ist.[42] Durch die Geometrie in Cycloalkinen ist die Dreifachbindung nicht mehr symmetrisch; stattdessen ergeben sich Angriffsvektoren orthogonal zur Ringebene, von außerhalb des Rings, sowie von innerhalb des Rings, wodurch auch transannulare Reaktionen möglich sind.[48] Beispielsweise kann in 5-Cyclodecinon das Sauerstoffatom der Carbonylgruppe unter Säurekatalyse die Dreifachbindung nucleophil angreifen, wodurch nach einer Umlagerung Bicyclo[4.4.0]-1(6)-decen-2-on entsteht.[144]
Reaktionen terminaler Alkine
[Bearbeiten | Quelltext bearbeiten]Terminale Alkine, also solche, die an einem Kohlenstoffatom der Dreifachbindung ein Wasserstoffatom tragen, gehen einige Reaktionen ein, die mit anderen Alkinen nicht möglich sind.
Sonogashira-Kupplung
[Bearbeiten | Quelltext bearbeiten]Die im Jahr 1975 entdeckte Sonogashira-Kupplung ist eine palladium-katalysierte Kupplungsreaktion, die die Kupplung eines terminalen Alkins mit einem Arylhalogenid oder Vinylhalogenid ermöglicht, und hat in der organischen Synthese eine große Bedeutung zur Knüpfung von C–C-Bindungen.[145][146] Als Edukte eignen sich Iodide, Bromide und Chloride, aber auch Triflate.[145] Als Produkte werden Arylalkine oder Enine erhalten.[146] Als Katalysator dienen Palladium(0)-spezies, die in situ erzeugt werden, zum Beispiel aus Tetrakis(triphenylphosphin)palladium(0) oder Bis(triphenylphosphin)palladium(II)-chlorid.[145] Zusätzlich wird eine Base benötigt, die jedoch in vielen Fällen auch direkt als Lösungsmittel verwendet werden kann.[145][146] In dieser Variante benötigt die Reaktion oft höhere Temperaturen bis zu 100 °C.[146] Durch Zusatz eines Kupfer-Katalysators (z. B. Kupfer(I)-iodid) ist die Reaktion jedoch auch bei Raumtemperatur möglich, dann muss allerdings unter Luftausschluss gearbeitet werden, um eine Glaser-Kupplung als Nebenreaktion zu vermeiden.[145][146] Die gewonnenen Arylalkine können synthetisch verschiedentlich weiterverwendet werden, zum Beispiel durch Hydrierung zu Alkanen und Alkenen oder zur Bildung von Heterocyclen (wie Benzofuranen, Indolen und Isochinolinen).[147] Die Sonogashira-Kupplung wurde auch zur Synthese vieler Naturstoffe verwendet[147] wie Desmosin[148], Pyrrhoxanthin[149][S 5] und Calicheamicin γ1.[147]
Glaser-Kupplung und verwandte Reaktionen
[Bearbeiten | Quelltext bearbeiten]Carl Glaser entdeckte 1869 in Bonn, dass Phenylacetylen unter Einwirkung von Kupfer(I)-chlorid und Luftsauerstoff zu Diphenyldiacetylen dimerisiert.[7] Die Glaser-Kupplung, bei der unter Kupfer-Katalyse und Einwirkung von Luftsauerstoff zwei terminale Alkine zu einem 1,3-Diin gekoppelt werden, war eine der ersten metall-katalysierten Kupplungsreaktionen.[150][151] Verwendet wurde die Glaser-Kupplung beispielsweise in einer frühen Synthese des Indigo.[7] Diese Reaktion wurde später weiterentwickelt: 1956 beschrieb Eglinton eine effizientere Reaktion mit Kupfer(II)-acetat und Pyridin in Methanol. 1962 beschrieb Hay eine Reaktion unter Verwendung von Kupfer(I)-chlorid und TMEDA an Luft. TMEDA verbessert die Löslichkeit der beteiligten Kupferverbindungen und damit das Reaktionsergebnis.[7][152][153] Eine solche modifizierte Reaktion wird daher auch als Glaser-Eglinton-Hay-Reaktion bezeichnet.[153] Inzwischen wurden diverse ähnliche Reaktionen mit anderen Katalysatormetallen beschrieben, darunter Palladium, Nickel, Silber und Gold.[154] Glaser-Kupplungen eignen sich zum Beispiel für die Synthese molekularer Knoten[155] und anderer makrocyclischer Verbindungen.[98]
Bei der Cadiot-Chodkiewicz-Reaktion wird ein terminales Alkin mit einem Bromalkin zu einem Diin umgesetzt. Dabei kommt Kupfer(I) (i. d. R. als Halogenid) zum Einsatz, sowie eine Base (z. B. Ethylamin), um entstehenden Bromwasserstoff abzufangen. Zusätzlich kann Hydroxylaminhydrochlorid als Reduktionsmittel zugesetzt werden, das eine oxidative Homokupplung verhindert.[156] Bei der Stephens-Castro-Kupplung wird ein Kupferacetylid mit einem Arylhalogenid oder Vinylhalogenid gekuppelt. Wird ein Arylhalogenid mit einem Stickstoff- oder Sauerstoffatom in ortho-Position verwendet (z. B. 2-Iodanilin), erfolgt eine Cyclisierung zu einem Indol oder Benzofuran.[157]
Click-Reaktionen
[Bearbeiten | Quelltext bearbeiten]Die kupfer(I)-katalysierte Umsetzung eines terminalen Alkins mit einem Azid zu einem Triazol, eine Variante der Huisgen-Reaktion, die auch als Sharpless-Click-Reaktion bezeichnet wird, ist die prototypische Reaktion der Click-Chemie.[158][159][160] Kupfer(I) kann in situ aus Kupfer(II)-sulfat und einem Reduktionsmittel erzeugt werden.[19] Die Edukte, Azide und Alkine sind leicht darstellbar, kinetisch stabil und vertragen sich mit vielen funktionellen Gruppen und Reaktionsbedingungen.[158] Die Reaktion verläuft in der Regel hochselektiv, praktisch quantitativ (typischerweise über 95 % Umsatz) und erfordert keine aufwendige Aufarbeitung.[158][159] Die Reaktion läuft außerdem bei niedrigen Temperaturen ab, oft bei Raumtemperatur, und kann in Wasser als Lösungsmittel durchgeführt werden.[159][160]
Durch ihre vorteilhaften Eigenschaften spielt diese Reaktion eine wichtige Rolle in bioorthognaler Chemie, bei der Reaktionen an Biomolekülen mit minimaler Interferenz durchgeführt werden können. Sie ermöglichen die Verkettung von Biomolekülen (Peptide, Proteine, Kohlenhydrate) oder deren Modifikation mit Flurophoren, Chlelatoren oder Isotopenmarkierungen. Dadurch ist auch das Labeling von Biomolekülen in vivo möglich.[158] Auch im Bereich der Materialwissenschaften findet die Technik Verwendung: Beispielsweise konnte eine mit 11-Azidoundecanthiol[S 6] funktionalisierte Goldoberfläche mit Ethinylferrocen[S 7] weiterfunktionalisiert werden. Sie ist außerdem in der supramolekularen Chemie und zur Funktionalisierung von Polymeren von Bedeutung.[159] Click-Reaktionen eignen sich auch für die kombinatorische Synthese von Molekülbibliotheken für die pharmazeutische Forschung.[160]
Halogenierung terminaler Alkine
[Bearbeiten | Quelltext bearbeiten]Acetylide können mit verschiedenen Reagenzien wie Chlor, Brom, Iod, Bromcyan, Iodcyan, N-Chlorsuccinimid oder N-Bromsuccinimid halogeniert werden. Mit Natriumhypochlorit oder Natriumhypobromit ist eine direkte Halogenierung terminaler Alkine möglich.[156]
Terminale Alkine als Nucleophile
[Bearbeiten | Quelltext bearbeiten]
Terminale Alkine sind Nucleophile, die an verschiedene Carbonyl- und Carboxylverbindungen addiert werden können. Eine Addition an ein Aldehyd ist zum Beispiel durch einen Silberkatalysator (Tricyclohexylphosphinsilber(I)-chlorid) möglich oder durch einen Mischkatalysator aus Ruthenium(III)-chlorid und Indium(III)-acetat.[161] Eine weitere Alternative ist die Katalyse mit Zinktriflat.[19] Die Addition an Säurechloride kann zum Beispiel mit einem Palladium-Kupfer-Katalysatorsystem durchgeführt werden. Auch Additionen an Imine und Iminium-Ionen, sowie 1,4-Additionen (z. B. an Vinylketone) sind möglich.[161]
Durch starke Basen können terminale Alkine quantitativ deprotoniert werden.[156] Möglich ist dies beispielsweise mit Lithiumalkylen (wie Butyllithium), mit Natriumamid, mit Lithiumdiisopropylamid oder mit Grignard-Verbindungen (wie Ethylmagnesiumbromid).[18][19] Mit den meisten Basen läuft die Deprotonierung schnell ab, mit Grignardverbindungen dauert sie hingegen länger.[156] Auch eine Deprotonierung mit elementaren Alkalimetallen ist möglich, wobei Wasserstoff-Gas als Nebenprodukt anfällt.[20] In deprotonierter Form können terminale Alkine leicht alkyliert werden, beispielsweise mit Epoxiden (wie Ethylenoxid), Aldehyden und Ketonen, sowie Alkylhalogeniden (wie Alkylbromiden).[18][19][20] Die Alkylierung funktioniert allerdings im Falle der Alkylhalogenide meistens nur mit Methylhalogeniden und primären Alkylhalogeniden, da das Alkinyl-Anion gegenüber sterisch anspruchsvolleren sekundären und tertiären Alkylhalogeniden als Base wirkt.[18] Liegt das Alkin jedoch in Form eines Trialkinylaluminiumreagenzes vor, ist hingegen auch eine Alkylierung mit sekundären und tertiären Alkylresten möglich.[156]
Kupfer- und Silber-Alkinylide können durch Umsetzung terminaler Alkine mit den Ammoniak-Komplexen von Kupfer(I)-chlorid oder Silbernitrat gewonnen werden. Sie fallen dabei als roter bzw. weißer Niederschläge aus, was sich als Nachweisreaktion eignet. Für synthetische Anwendungen sind sie aufgrund ihrer Brisanz weniger geeignet.[20]
Die Umsetzung von Acetylen mit einem Überschuss Kaliumhydroxid und einem Keton zu einem Propargylalkohol wird als Favorski-Reaktion bezeichnet.[162]
Weitere Reaktionen terminaler Alkine
[Bearbeiten | Quelltext bearbeiten]Terminale Alkine sind auch Elektrophile.[18][19] Durch Aktivierung mit einer Gold(I)- oder Gold(III)-verbindung (z. B. Gold(I)-chlorid) können Nukleophile wie Wasser an terminale Alkine addiert werden.[19] Auch Normant-Cuprate können (selbst ohne Aktivierung) an terminale Alkine addiert werden, wobei Alkenylkupfer-Spezies entstehen. Diese können durch saure Aufarbeitung protoniert oder mit anderen Elektrophilen umgesetzt werden, beispielsweise mit Iod, Chlorcyan oder Ethylenoxid.[103]
Durch Umsetzung mit Silbercarbonat und Trimethylsilylazid können terminale Alkine in Nitrile überführt werden.[163] Durch Umsetzung mit katalytisch Kalium-tert-butanolat können terminale Alkine (1-Alkine) in interne Alkine (2-Alkine) isomerisiert werden. Auch Isomerisierungen zu Allenen sind bekannt, diese haben jedoch meist keine synthetische Bedeutung, weil sie schlecht zu trennende Gemische von Produkten ergeben.[156]
Schutzgruppen für terminale Alkine
[Bearbeiten | Quelltext bearbeiten]Terminale Alkine können mit einer Trimethylsilylgruppe geschützt werden.[19][156] Hierfür wird die Verbindung zunächst mit einer starken Base deprotoniert und dann mit Trimethylsilylchlorid umgesetzt.[19] Trimethylsilyl- und Trimethylgermanylgruppen eignen sich als orthogonale Schutzgruppen, da erstere selektiv mit Kaliumfluorid und [18]Krone-6 und letztere selektiv mit Kupfer(I)-bromid entschützt werden kann.[164] Eine alternative Schutzgruppe ist ein Diphenylphosphinoxid, das durch Umsetzung mit Chlordiphenylphosphin und Oxidation mit Wasserstoffperoxid eingeführt und mit Kalium-tert-butanolat entfernt werden kann.[165]
Alkine als Komplexliganden
[Bearbeiten | Quelltext bearbeiten]Alkine bilden Komplexverbindungen mit verschiedenen Übergangsmetallen in diversen Bindungsmodi, was oft auch einen erheblichen Einfluss auf die Bindungssituation im Liganden hat, beispielsweise eine Abweichung des Bindungswinkels von der Linearität.[48] Acetylid-Ionen bilden Komplexe unter anderem mit Mangan, Eisen, Cobalt, Nickel und Kupfer. Diese weisen überwiegend dieselben Stöchiometrien, Farben und magnetischen Eigenschaften auf wie die analogen Cyanid-Komplexe, sind aber explosiv und sehr pyrolyseempfindlich.[166] Von Platin ist eine große Anzahl an Komplexen mit Alkinliganden bekannt, darunter sowohl η1- als auch η2-Komplexe.[167] Auch Rhodium-Alkin-Komplexe sind bekannt.[168] Durch Komplexierung können stark gespannte Cycloalkine stabilisiert werden. Cyclopentin, Cyclohexin und Cycloheptin sind als freie Verbindungen nicht isolierbar, sind aber als Liganden von Übergangsmetallen wie Zirconium, Nickel, Platin oder Osmium bekannt.[37] Beispielsweise können ausgehend von 1-Bromcyclohexen und 1-Bromcyclohepten durch Eliminierung mit Lithiumdiisopropylamid in Gegenwart von Tris(triphenylphosphin)platin(0) die entsprechenden Cycloalkin-Platinkomplexe gewonnen werden.[169] Auch durch Reduktion von 1,2-Dibromcyclopenten und 1,2-Dibromcyclohexen mit Natriumamalgam in Gegenwart dieses Platinkomplexes, können entsprechende Komplexe der Cycloalkine erzeugt werden.[170]
Bedeutung und Verwendung
[Bearbeiten | Quelltext bearbeiten]Von großer technischer Bedeutung sind unter den Alkinen lediglich Acetylen und Propin. Etwa drei Viertel dieser beiden Verbindungen werden zur Synthese, vor allem für Polymerisationsreaktionen, eingesetzt.[171] Insbesondere die Monomere Vinylchlorid, Vinylacetat und Acrylsäure werden z. T. noch aus Acetylen hergestellt, obwohl ein Großteil der Weltproduktion dieser Verbindungen heutzutage aus billigerem Ethylen (bzw. Propylen im Fall der Acrylsäure) hergestellt wird. Der größte Produzent von Acetylen (sowie mit 80 % der weltweiten Produktion der größte Verbraucher) ist China, wo nach 2010 noch neue Anlagen zur Herstellung von Acetylen über Calciumcarbid in Betrieb genommen wurden.[9] Bei der Produktion von anderen Alkinen ist Acetylen nach wie vor die wichtigste Ausgangsverbindung, z. B. bei Butindiol, das selbst wiederum ein wichtiger Vorläufer für die Herstellung von Tetrahydrofuran (THF) ist.[9][172]
Acetylen-Sauerstoff-Flammen werden mit bis zu 3100 °C extrem heiß, weshalb Acetylen für Schweiß- und Schneidbrenner verwendet wird.[9][171] Je nach Verhältnis von Sauerstoff zu Acetylen ergibt sich eine oxidierende Flamme (z. B. zum Schweißen von Messing, sowie zum Schneiden von Werkstoffen und Härten von Oberflächen) oder eine reduzierende Flamme (z. B. zum Schweißen von Stahl, Kupfer und Aluminium).[9]
Aufgrund der Giftigkeit von Hydrazin werden verstärkt weniger bedenkliche Raketentreibstoffe erforscht. Auch ein hypergoles Gemisch aus Propin und Wasserstoffperoxid ist ein Kandidat hierfür.[173] Acetylen wirkt auf Pflanzen ähnlich wie das Reifungshormon Ethylen, daher wird aus Calciumcarbid erzeugtes Acetylen teilweise als Reifungsbeschleuniger verwendet, z. B. im Vertrieb von Bananen.[174][175]
-
 Die Monomere diverser technischer Polymere werden aus Acetylen hergestellt, zum Beispiel Vinylchlorid als Monomer für Polyvinylchlorid, aus dem z. B. Rohre produziert werden
Die Monomere diverser technischer Polymere werden aus Acetylen hergestellt, zum Beispiel Vinylchlorid als Monomer für Polyvinylchlorid, aus dem z. B. Rohre produziert werden -
 THF wird ausgehend von Acetylen produziert
THF wird ausgehend von Acetylen produziert -
 Durch Verbrennung von Acetylen kann eine sehr heiße und helle Flamme erzeugt werden, die z. B. zum Schweißen verwendet wird
Durch Verbrennung von Acetylen kann eine sehr heiße und helle Flamme erzeugt werden, die z. B. zum Schweißen verwendet wird -
 Acetylen wird als Reifungsbeschleuniger verwendet, z. B. für Bananen
Acetylen wird als Reifungsbeschleuniger verwendet, z. B. für Bananen




Alkine in der Medizin
[Bearbeiten | Quelltext bearbeiten]Ethinylgruppen dienen im Wirkstoffdesign verschiedenen Zwecken. Eine solche Dreifachbindung verhält sich isoster zu diversen anderen Gruppen. Hinsichtlich der π-Elektronen weist sie Ähnlichkeit mit sechsgliedrigen oder auch kleineren aromatischen Ringen auf. Elektrostatisch verhält sich die Ethinylgruppe ähnlich zu Chlor-, Brom- oder Iodsubstituenten und ist bei letzterem auch in der Länge ähnlich. Diese Alkingruppen ermöglichen die Steuerung von Moleküleigenschaften und dadurch indirekt die von pharmakologischen Parametern wie Bioverfügbarkeit, Target-Selektivität und metabolische Stabilität von Wirkstoffen. Die Ethinylgruppe ist außerdem geometrisch ähnlich zur Nitrilgruppe. Allerdings ist gegenüber dieser einerseits die elektronenziehende Eigenschaft der Ethinylgruppe schwächer ausgeprägt[10] sowie andererseits die Elektronendichte am Wasserstoffatom geringer.
Terminale, aber auch internale Dreifachbindungen eignen sich als Warhead für kovalente Inhibitoren. Eine terminale Dreifachbindung ist in der Regel mäßig reaktiv, was unerwünschte Nebenreaktionen gegenüber reaktivieren Gruppen reduziert. Trotzdem treten diese zum Teil auf und betreffen dann die Konjugation mit Glutathion und die irreversible Hemmung von Cytochrom-P450-Enzymen. Da diese Enzyme eine wichtige Rolle bei der Metabolisierung von Arzneistoffen spielen, kann ihre Ausschaltung zu problematischen Wechselwirkungen mit anderen Arzneistoffen führen, soweit kein angemessener zeitlicher Abstand bei ihrer Einnahme eingehalten wird.[10]
Verschiedene Alkine werden als Arzneistoffe verwendet. Viele darunter sind synthetische Steroidhormonderivate, die für die Kontrazeption und Hormonersatztherapie verwendet werden. Zu ihnen gehören Estrogene wie das Ethinylestradiol – eines der meistgenutzten Kontrazeptiva – und sein Prodrug Mestranol.[176] Sie tragen wie auch manche synthetische Gestagene (etwa Norethindron,[177] Gestoden, Desogestrel[178] Levonorgestrel,[179] Etonogestrel,[180] Etynodioldiacetat[181] Norgestimat[182][S 8] und sein Metabolit Norelgestromin[183][S 9]) sowie der zum medikamentösen Schwangerschaftsabbruch verwendete Progesteronrezeptor-Antagonist Mifepriston[184] am C-17 des Steroidgerüsts eine Ethinylgruppe, die die rasche Inaktivierung des jeweiligen Stoffes in der Leber verhindert.[185] Die 17-OH-Gruppe derartiger Verbindungen kann normalerweise leicht zum Keton oxidiert werden, was im Allgemeinen einen deutlichen Wirkungsverlust bedeutet. Die Ethinylgruppe verhindert diese Oxidation jedoch.[10] Dies ist auch der Fall bei Danazol, das bei Endometriose eingesetzt wird.[10][186]
Selegilin und Rasagilin werden als Inhibitoren der Monoaminoxidase B bei Parkinson-Patienten eingesetzt.[187][188] Die Wirkung beruht auf der Ausbildung einer kovalenten Bindung über die Ethinylgruppe der Wirkstoffe mit einer Struktur im Flavin-Adenin-Dinukleotid(FAD)-Cofaktor des Enzyms, was zu einer irreversiblen Hemmung führt.[10][189] Tremorin[S 10] wirkt krampfauslösend und wird bei Tierversuchen in der Parkinsonforschung verwendet.[190]
Mehrere Pharmazeutika mit Dreifachbindungen werden bei Krebserkrankungen eingesetzt. Dazu gehört der bei Lungenkrebs verwendete Tyrosinkinasehemmer Erlotinib, das als Inhibitor des Rezeptors für den epidermalen Wachstumsfaktor (EGF-Rezeptor, EGFR) wirkt,[191][192] indem er an die ATP-Bindungsstelle in der mutierten Kinasedomäne des EGFR bindet.[193] Der Wirkstoff geht jedoch unerwünschte Nebenreaktionen mit mehreren Cytochrom-P450-Enzymen ein, insbesondere CYP3A4. Einerseits kann die Verbindung einige der Enzyme irreversibel hemmen, andererseits kann die Dreifachbindung durch Oxidation in ein hochreaktives Oxiren oder Keten umgewandelt werden.[194] Futibatinib, das im September 2022 in den USA zugelassen wurde, ist ein Krebsmedikament für verschiedene Arten von Krebs, u. a. für Brustkrebs, Magenkrebs, Speiseröhrenkrebs und Lungenkrebs.[195] Die systemische Toxizität mancher cytotoxischer Wirkstoffe kann durch das Konzept des Drug-Targeting reduziert werden. Durch Kopplung an einen Antikörper entsteht ein Immunkonjugat (Antibody Drug Conjugate, ADC), das die zielgerichtete Behandlung von bestimmten Krebserkrankungen ermöglicht. Zwei Beispiele sind das gegen akute lymphatische Leukämie (ALL) zugelassene Gemtuzumab-Ozogamicin (USA, 2000, 2017)[196] und das gegen akute myeloische Leukämie (AML) zugelassene Inotuzumab-Ozogamicin (EU, 2017).[196][197] Bei den beiden ADC ist der eigentliche Wirkstoff („Payload“) das halbsynthetische Derivat eines hochtoxischen Bakteriengifts aus der Gruppe der Calicheamicine, das eine Endiin-Substruktur enthält.[196] Dieses kann durch Bergman-Cyclisierung und Bildung eines Diradikals einen Doppelstrangbruch in der DNA und dadurch eine Apoptose (Zelltod) der betroffenen Zelle verursachen.[196] Die Einschleusung des Wirkstoffs in die Zellen funktioniert durch die Bindung des Antikörpers an bestimmte Rezeptoren. Der monoklonales Antikörper Gemtuzumab bindet beispielsweise an den Rezeptor CD33, während der monoklonale Antikörper Inotuzumab an den Rezeptor CD22 bindet.[198]
-
 Die Einführung einer Ethinylgruppe am C-17 von Steroidhormonen (z. B. Ethinylestradiol) verlangsamt deren metabolischen Abbau
Die Einführung einer Ethinylgruppe am C-17 von Steroidhormonen (z. B. Ethinylestradiol) verlangsamt deren metabolischen Abbau -
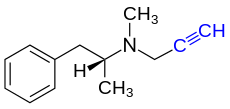 Die endständige Alkingruppe des Parkinsonmittels Selegilin bindet kovalent an Strukturen eines Enzym-Cofaktors
Die endständige Alkingruppe des Parkinsonmittels Selegilin bindet kovalent an Strukturen eines Enzym-Cofaktors -
 Das Prodrug Tazaroten hat durch die interne Alkinylstruktur eine starre Konformation, auf der die hohe Selektivität des wirksamen Metaboliten für bestimmte Retinoidrezeptoren beruht
Das Prodrug Tazaroten hat durch die interne Alkinylstruktur eine starre Konformation, auf der die hohe Selektivität des wirksamen Metaboliten für bestimmte Retinoidrezeptoren beruht -
 Dreifachbindungen im reaktiven Endiin-Strukturelement des Bakterientoxins Calicheamicin γ1
Dreifachbindungen im reaktiven Endiin-Strukturelement des Bakterientoxins Calicheamicin γ1



diin-substructure.svg)
Pralatrexat wird bei entzündlichen Erkrankungen verwendet.[199] Tazaroten ist ebenfalls ein entzündungshemmender Wirkstoff, der an die Retinoid-Rezeptoren RARβ und RARγ bindet, und wird insbesondere bei Akne und Schuppenflechte eingesetzt.[10][200] In diesem Fall führte der Einbau einer Dreifachbindung durch die starrere Konformation zu einer besseren Selektivität gegenüber diesen spezifischen Rezeptoren.[10]
Efavirenz ist ein HIV-1-Medikament.[201] Lenacapavir, das ebenfalls gegen HIV verwendet wird, wurde von Gilead Sciences entwickelt und wurde 2022 als erster Kapsidinhibitor in den USA, Kanada und in der EU zugelassen.[202] Haloprogin ist ein topisches Fungizid[203] und Terbinafin wird bei Mykosen eingesetzt.[204] Das Anticholinergikum Oxybutynin wird bei übermäßigem Harndrang angewendet,[205] das Prostacyclinanalogon Iloprost bei pulmonaler Hypertonie.[206] Das Schlafmittel Ethinamat ist nicht mehr im Handel.[207] Linagliptin, das erst im September 2022 in den USA zugelassen wurde, findet Verwendung bei der Behandlung von Diabetes mellitus Typ 2.[208]
Alkine in der Schädlingsbekämpfung
[Bearbeiten | Quelltext bearbeiten]

Als Herbizide werden diverse Verbindungen verwendet, die als Inhibitoren der Protoporphyrinogen-IX-Oxidase wirken. Ihre Wirkung besteht in der Akkumulation von Protoporphyrin IX durch die Enzymhemmung. Dieses ist ein starker Photosensibilisator, der unter Einfluss von Sonnenlicht größere Mengen Singulett-Sauerstoff erzeugt, der zur Schädigung und zum Absterben von Blättern führt.[209][210][211] Viele Wirkstoffe dieses Typs tragen eine Propargylgruppe. Diese ist vermutlich wichtig für die Bindungseigenschaften, da sie den Vinylgruppen des Protoporphyrin IX ähnelt, das das natürliche Substrat des Enzyms ist.[209] Zu diesen Verbindungen gehört beispielsweise das Thidiazimin, das gegen verschiedene Unkräuter wirkt, jedoch unschädlich für verschiedene Getreidearten wie Triticale oder Winterweizen ist.[212] Weitere Vertreter sind Oxadiargyl, Pyraclonil, Flumioxazin, Flumipropyn und Azafenidin. Kommerzielle Herbizide, die ebenfalls Alkine sind, jedoch eine andere Wirkungsweise haben, sind beispielsweise Clodinafop, Propyzamid und Prynachlor.[209]
Zu den Alkinen, die als Fungizide verwendet werden, gehört das Mepanipyrim[209], das die Methionin-Biosynthese inhibiert, sowie das Capillin.[213] Mandipropamid wirkt gegen verschiedene Eipilze und wird ausgehend von 4-Hydroxy-3-methoxyphenethylamin synthetisiert. Die Propargylgruppe wird mit Propargylbromid eingeführt.[214]
Viele insektizide Ester der Chrysanthemumsäure wie die Allethrine tragen eine Allylgruppe. Solche, die stattdessen eine Propargylgruppe tragen, sind jedoch ähnlich oder mehr wirksam.[215] Kommerziell genutzte Vertreter dieser Gruppe, die eine Ethinyl- oder Propargylgruppe tragen, sind Prallethrin, Imiprothrin, Furamethrin, Kikuthrin und Empenthrin.[209]
Literatur
[Bearbeiten | Quelltext bearbeiten]- Peter J. Stang, François Diederich: Modern acetylene chemistry. VCH, Weinheim 1995, ISBN 3-527-29084-2.
- Lambert Brandsma: Synthesis of acetylenes, allenes and cumulenes (= Best synthetic methods). Elsevier, Amsterdam 2004, ISBN 0-12-125751-7.
- K. Peter C. Vollhardt, Neil E. Schore, Übersetzung herausgegeben von Holger Butenschön: Organische Chemie. Weinheim 2020, ISBN 978-3-527-34582-3, S. 663–677.
Alkine als Naturstoffe:
- Dmitry V. Kuklev, Abraham J. Domb, Valery M. Dembitsky: Bioactive acetylenic metabolites. In: Phytomedicine. Band 20, Nr. 13, Oktober 2013, S. 1145–1159, doi:10.1016/j.phymed.2013.06.009.
- Robert E. Minto, Brenda J. Blacklock: Biosynthesis and function of polyacetylenes and allied natural products. In: Progress in Lipid Research. Band 47, Nr. 4, Juli 2008, S. 233–306, doi:10.1016/j.plipres.2008.02.002.
Weblinks
[Bearbeiten | Quelltext bearbeiten]Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ a b Barry M. Trost and Chao-Jun Li: Modern Alkyne Chemistry. Wiley, ISBN 978-3-527-33505-3, S. 1 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Jaime Wisniak: Edmond Davy. In: Educación Química. Band 31, Nr. 4, 6. Oktober 2020, S. 144, doi:10.22201/fq.18708404e.2020.4.72934.
- ↑ Justin Russell: Edmund Davy. In: Journal of Chemical Education. Band 30, Nr. 6, Juni 1953, S. 302, doi:10.1021/ed030p302.
- ↑ Robert E. Minto, Brenda J. Blacklock: Biosynthesis and function of polyacetylenes and allied natural products. In: Progress in Lipid Research. Band 47, Nr. 4, Juli 2008, S. 233–306, doi:10.1016/j.plipres.2008.02.002, PMID 18387369, PMC 2515280 (freier Volltext).
- ↑ Jesse A. Grantham, Richard W. McLay: Engineering Standards in Weld Failure. In: Engineering Standards for Forensic Application. Elsevier, 2019, ISBN 978-0-12-813240-1, S. 189–198, doi:10.1016/b978-0-12-813240-1.00014-5.
- ↑ a b c d Åshild Moi Sørskår, Helge Ø. K. Stenstrøm, Yngve Stenstrøm, Simen Gjelseth Antonsen: The Alkyne Zipper Reaction: A Useful Tool in Synthetic Chemistry. In: Reactions. Band 4, Nr. 1, 30. Dezember 2022, S. 26–65, doi:10.3390/reactions4010002.
- ↑ a b c d Peter Siemsen, Robert C. Livingston, François Diederich: Acetylenic Coupling: A Powerful Tool in Molecular Construction. In: Angewandte Chemie International Edition. Band 39, Nr. 15, 4. August 2000, S. 2632–2657, doi:10.1002/1521-3773(20000804)39:15<2632::AID-ANIE2632>3.0.CO;2-F.
- ↑ Yahraes, Herbert. “THE ARRIVAL OF ACETYLENE.” Scientific American, vol. 180, no. 1, 1949, pp. 16–21
- ↑ a b c d e Ullmann's Encyclopedia of Industrial Chemistry. 1. Auflage. Wiley, 2003, ISBN 3-527-30385-5, doi:10.1002/14356007.a01_097.pub4.
- ↑ a b c d e f g h Tanaji T. Talele: Acetylene Group, Friend or Foe in Medicinal Chemistry. In: Journal of Medicinal Chemistry. Band 63, Nr. 11, 11. Juni 2020, S. 5625–5663, doi:10.1021/acs.jmedchem.9b01617.
- ↑ Floyd L. Klavetter, Robert H. Grubbs: Polycyclooctatetraene (polyacetylene): synthesis and properties. In: Journal of the American Chemical Society. Band 110, Nr. 23, November 1988, S. 7807–7813, doi:10.1021/ja00231a036.
- ↑ a b c Hideki Shirakawa: The Discovery of Polyacetylene Film: The Dawning of an Era of Conducting Polymers (Nobel Lecture). In: Angewandte Chemie International Edition. Band 40, Nr. 14, 16. Juli 2001, S. 2574–2580, doi:10.1002/1521-3773(20010716)40:14<2574::AID-ANIE2574>3.0.CO;2-N.
- ↑ C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau, Alan G. MacDiarmid: Electrical Conductivity in Doped Polyacetylene. In: Physical Review Letters. Band 39, Nr. 17, 24. Oktober 1977, S. 1098–1101, doi:10.1103/PhysRevLett.39.1098.
- ↑ Hans-Georg Elias: Makromoleküle. 4: Anwendungen von Polymeren. 6. vollst. überarb. Auflage. Wiley-VCH, Weinheim 2003, ISBN 3-527-29962-9.
- ↑ Bernd Tieke: Makromolekulare Chemie: eine Einführung. Dritte Auflage. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 2014, ISBN 978-3-527-33216-8.
- ↑ The Nobel Prize in Chemistry 2000. Abgerufen am 12. September 2023 (amerikanisches Englisch).
- ↑ The Nobel Prize in Chemistry 2022. Abgerufen am 9. September 2023 (amerikanisches Englisch).
- ↑ a b c d e f g h i j k l m n o p q r s K. Peter C. Vollhardt, Neil E. Schore, Übersetzung herausgegeben von Holger Butenschön: Organische Chemie. Weinheim 2020, ISBN 978-3-527-34582-3, S. 663–677
- ↑ a b c d e f g h i j k l m n o p q r Jonathan Clayden, Nick Greeves, Stuart G. Warren: Organische Chemie (= Lehrbuch). 2. Auflage [2., korrigierter Nachdruck]. Springer Spektrum, Berlin 2017, ISBN 978-3-642-34715-3.
- ↑ a b c d e f g h i Paul Arnaud, Brigitte Jamart, Jacques Bodiguel, Nicolas Brosse: Chimie organique: les cours de Paul Arnaud avec 350 questions et exercices corrigés (= Sciences Sup). 18e éd. entièrement refondue. Dunod, Paris 2009, ISBN 978-2-10-052647-5.
- ↑ a b Eintrag zu Propin in der GESTIS-Stoffdatenbank des IFA, abgerufen am 8. Januar 2021. (JavaScript erforderlich)
- ↑ a b Eintrag zu 1-Butin in der GESTIS-Stoffdatenbank des IFA, abgerufen am 8. Januar 2021. (JavaScript erforderlich)
- ↑ a b Eintrag zu 2-Butin in der GESTIS-Stoffdatenbank des IFA, abgerufen am 8. Januar 2021. (JavaScript erforderlich)
- ↑ a b Datenblatt 1-Pentin bei Merck, abgerufen am 13. März 2023.
- ↑ a b Datenblatt 2-Pentin bei Merck, abgerufen am 21. Februar 2010.
- ↑ a b c d e f David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press / Taylor and Francis, Boca Raton FL, Physical Constants of Organic Compounds, S. 3-284.
- ↑ a b David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press / Taylor and Francis, Boca Raton FL, Physical Constants of Organic Compounds, S. 3-276.
- ↑ a b c David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 96. Auflage. CRC Press / Taylor and Francis, Boca Raton FL, Physical Constants of Organic Compounds, S. 3-292.
- ↑ a b c d e f g h David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press / Taylor and Francis, Boca Raton FL, Physical Constants of Organic Compounds, S. 3-406.
- ↑ a b c d A M Sladkov, Yu P Kudryavtsev: POLYYNES. In: Russian Chemical Reviews. Band 32, Nr. 5, 31. Mai 1963, S. 229–243, doi:10.1070/RC1963v032n05ABEH001338.
- ↑ a b S A Hutchinson: Biological Activities of Volatile Fungal Metabolites. In: Annual Review of Phytopathology. Band 11, Nr. 1, September 1973, S. 223–246, doi:10.1146/annurev.py.11.090173.001255.
- ↑ Donald W. Rogers, Nikita Matsunaga, Andreas A. Zavitsas, Frank J. McLafferty, Joel F. Liebman: The Conjugation Stabilization of 1,3-Butadiyne Is Zero. In: Organic Letters. Band 5, Nr. 14, 1. Juli 2003, S. 2373–2375, doi:10.1021/ol030019h.
- ↑ a b c Yueze Gao, Rik R. Tykwinski: Advances in Polyynes to Model Carbyne. In: Accounts of Chemical Research. Band 55, Nr. 24, 20. Dezember 2022, S. 3616–3630, doi:10.1021/acs.accounts.2c00662.
- ↑ Ray H. Baughman: Dangerously Seeking Linear Carbon. In: Science. Band 312, Nr. 5776, 19. Mai 2006, S. 1009–1110, doi:10.1126/science.1125999.
- ↑ Ferdinand Bohlmann: Polyacetylene, IV. Mitteil.: Darstellung von Di‐ tert .‐butyl‐polyacetylenen. In: Chemische Berichte. Band 86, Nr. 5, Mai 1953, S. 657–667, doi:10.1002/cber.19530860519.
- ↑ Wesley A. Chalifoux, Rik R. Tykwinski: Synthesis of polyynes to model the sp-carbon allotrope carbyne. In: Nature Chemistry. Band 2, Nr. 11, November 2010, S. 967–971, doi:10.1038/nchem.828.
- ↑ a b c Martin A. Bennett, Heinz P. Schwemlein: Metal Complexes of Small Cycloalkynes and Arynes. In: Angewandte Chemie International Edition in English. Band 28, Nr. 10, Oktober 1989, S. 1296–1320, doi:10.1002/anie.198912961.
- ↑ a b Dieter Heber, Peter Rösner, Werner Tochtermann: Cyclooctyne and 4‐Cyclooctyn‐1‐ol – Versatile Building Blocks in Organic Synthesis. In: European Journal of Organic Chemistry. Band 2005, Nr. 20, Oktober 2005, S. 4231–4247, doi:10.1002/ejoc.200500288.
- ↑ A. T. Blomquist, Liang Huang Liu: Many-membered Carbon Rings. VII. Cycloöctyne. In: Journal of the American Chemical Society. Band 75, Nr. 9, Mai 1953, S. 2153–2154, doi:10.1021/ja01105a039.
- ↑ a b L. Brandsma, H. D. Verkruijsse: An Improved Synthesis of Cyclooctyne. In: Synthesis. Band 1978, Nr. 04, 1978, S. 290–290, doi:10.1055/s-1978-24725.
- ↑ S. A. Krouse, R. R. Schrock, R. E. Cohen: Ring-opening polymerization of cyclooctyne. In: Macromolecules. Band 20, Nr. 4, April 1987, S. 903–904, doi:10.1021/ma00170a033.
- ↑ a b Kimberly Chenoweth, David Chenoweth, William A. Goddard III: Cyclooctyne-based reagents for uncatalyzed click chemistry: A computational survey. In: Organic & Biomolecular Chemistry. Band 7, Nr. 24, 2009, S. 5255, doi:10.1039/b911482c.
- ↑ John C. Gilbert, Everett G. McKinley, Duen-Ren Hou: The Nature of Cyclopentyne from Different Precursors. In: Tetrahedron. Band 53, Nr. 29, Juli 1997, S. 9891–9902, doi:10.1016/S0040-4020(97)00334-7.
- ↑ Christian M. Gampe, Erick M. Carreira: Arynes and Cyclohexyne in Natural Product Synthesis. In: Angewandte Chemie International Edition. Band 51, Nr. 16, 16. April 2012, S. 3766–3778, doi:10.1002/anie.201107485.
- ↑ Heather A. Carlson, Geoffrey E. Quelch, Henry F. Schaefer: How stable is cyclobutyne? The activation energy for the unimolecular rearrangement to butatriene. In: Journal of the American Chemical Society. Band 114, Nr. 13, Juni 1992, S. 5344–5348, doi:10.1021/ja00039a053.
- ↑ Zhi Sun, Henry F. Schaefer: Cyclobutyne: Minimum or Transition State? In: The Journal of Organic Chemistry. Band 84, Nr. 9, 3. Mai 2019, S. 5548–5553, doi:10.1021/acs.joc.9b00502.
- ↑ Herbert Meier, Norbert Hanold, Thomas Molz, Hans Joachim Bissinger, Heinz Kolshorn, Johannes Zountsas: Strained cycloalkenynes. In: Tetrahedron. Band 42, Nr. 6, Januar 1986, S. 1711–1719, doi:10.1016/S0040-4020(01)87588-8.
- ↑ a b c d Peter J. Stang, François Diederich: Modern acetylene chemistry. VCH, Weinheim 1995, ISBN 3-527-29084-2.
- ↑ a b c d e Dmitry V. Kuklev, Abraham J. Domb, Valery M. Dembitsky: Bioactive acetylenic metabolites. In: Phytomedicine. Band 20, Nr. 13, Oktober 2013, S. 1145–1159, doi:10.1016/j.phymed.2013.06.009.
- ↑ Samuele Sala, Jane Fromont, Oliver Gomez, Daniel Vuong, Ernest Lacey, Gavin R. Flematti: Albanitriles A–G: Antiprotozoal Polyacetylene Nitriles from a Mycale Marine Sponge. In: Journal of Natural Products. Band 82, Nr. 12, 27. Dezember 2019, S. 3450–3455, doi:10.1021/acs.jnatprod.9b00840.
- ↑ Edgar B. Cahoon, Judy A. Schnurr, Errol A. Huffman, Robert E. Minto: Fungal responsive fatty acid acetylenases occur widely in evolutionarily distant plant families: Fungal responsive fatty acid acetylenases. In: The Plant Journal. Band 34, Nr. 5, Juni 2003, S. 671–683, doi:10.1046/j.1365-313X.2003.01757.x.
- ↑ K. L. Mikolajczak, C. R. Smith, M. O. Bagby, I. A. Wolff: A New Type of Naturally Occurring Polyunsaturated Fatty Acid. In: The Journal of Organic Chemistry. Band 29, Nr. 2, Februar 1964, S. 318–322, doi:10.1021/jo01025a016.
- ↑ Kurt Aitzetmüller: Santalbic acid in the plant kingdom. In: Plant Systematics and Evolution. Band 298, Nr. 9, November 2012, S. 1609–1617, doi:10.1007/s00606-012-0678-5.
- ↑ Marcel S. F. Lie Ken Jie, Mohammed Khysar Pasha, Fasih Ahmad: Ultrasound-assisted synthesis of santalbic acid and a study of triacylglycerol species inSantalum album (Linn.) seed oil. In: Lipids. Band 31, Nr. 10, Oktober 1996, S. 1083–1089, doi:10.1007/BF02522466.
- ↑ D. A. Konovalov: Polyacetylene Compounds of Plants of the Asteraceae Family (Review). In: Pharmaceutical Chemistry Journal. Band 48, Nr. 9, Dezember 2014, S. 613–631, doi:10.1007/s11094-014-1159-7.
- ↑ Leo J. Schep, Robin J. Slaughter, Gordon Becket, D. Michael G. Beasley: Poisoning due to water hemlock. In: Clinical Toxicology. Band 47, Nr. 4, April 2009, S. 270–278, doi:10.1080/15563650902904332.
- ↑ Matthew Tcheng, Mark D. Minden, Paul A. Spagnuolo: Avocado‐derived avocadyne is a potent inhibitor of fatty acid oxidation. In: Journal of Food Biochemistry. Band 46, Nr. 3, März 2022, doi:10.1111/jfbc.13895.
- ↑ Alex Sinclair, Robert A. Stockman: Thirty-five years of synthetic studies directed towards the histrionicotoxin family of alkaloids. In: Natural Product Reports. Band 24, Nr. 2, 2007, S. 298, doi:10.1039/b604203c.
- ↑ a b John W. Blunt, Brent R. Copp, Murray H. G. Munro, Peter T. Northcote, Michèle R. Prinsep: Marine natural products. In: Natural Product Reports. Band 20, Nr. 1, 21. Januar 2003, S. 1–48, doi:10.1039/b207130b.
- ↑ John W. Blunt, Brent R. Copp, Wan-Ping Hu, Murray H. G. Munro, Peter T. Northcote, Mich?le R. Prinsep: Marine natural products. In: Natural Product Reports. Band 24, Nr. 1, 2007, S. 31, doi:10.1039/b603047p.
- ↑ John W. Blunt, Brent R. Copp, Wan-Ping Hu, Murray H. G. Munro, Peter T. Northcote, Michèle R. Prinsep: Marine natural products. In: Natural Product Reports. Band 26, Nr. 2, 2009, S. 170, doi:10.1039/b805113p.
- ↑ a b John W. Blunt, Brent R. Copp, Murray H. G. Munro, Peter T. Northcote, Michèle R. Prinsep: Marine natural products. In: Natural Product Reports. Band 21, Nr. 1, 2004, S. 1, doi:10.1039/b305250h.
- ↑ a b John W. Blunt, Brent R. Copp, Murray H. G. Munro, Peter T. Northcote, Michèle R. Prinsep: Marine natural products. In: Natural Product Reports. Band 22, Nr. 1, 2005, S. 15, doi:10.1039/b415080p.
- ↑ Walter D. Celmer, I. A. Solomons: Mycomycin. I. Isolation, Crystallization and Chemical Characterization. In: Journal of the American Chemical Society. Band 74, Nr. 9, Mai 1952, S. 2245–2248, doi:10.1021/ja01129a024.
- ↑ Qiu-Ye Chai, Zhen Yang, Hou-Wen Lin, Bing-Nan Han: Alkynyl-Containing Peptides of Marine Origin: A Review. In: Marine Drugs. Band 14, Nr. 11, 23. November 2016, S. 216, doi:10.3390/md14110216, PMID 27886049, PMC 5128759 (freier Volltext).
- ↑ a b Mukesh C. Joshi, Diwan S. Rawat: Recent Developments in Enediyne Chemistry. In: Chemistry & Biodiversity. Band 9, Nr. 3, März 2012, S. 459–498, doi:10.1002/cbdv.201100047.
- ↑ a b c Matija Gredičak, Ivanka Jerić: Enediyne compounds - new promises in anticancer therapy. In: Acta Pharmaceutica. Band 57, Nr. 2, 1. Juni 2007, S. 133–150, doi:10.2478/v10007-007-0011-y.
- ↑ Nakao Ishida, Keizō Miyazaki, Katsuo Kumagai, Mitsuo Rikimaru: Neocarzinostatin, an Antitumor Antibiotic of High Molecular Weight Isolation, Physicochemical Properties and Biological Activities. In: The Journal of Antibiotics, Series A. Band 18, Nr. 2, 1965, S. 68–76, doi:10.11554/antibioticsa.18.2_68.
- ↑ Nada Zein, Achyut M. Sinha, William J. McGahren, George A. Ellestad: Calicheamicin γ 1 I : an Antitumor Antibiotic That Cleaves Double-Stranded DNA Site Specifically. In: Science. Band 240, Nr. 4856, 27. Mai 1988, S. 1198–1201, doi:10.1126/science.3240341.
- ↑ Masataka Konishi, Hiroaki Ohkuma, Kyo-Ichiro Saitoh, Hiroshi Kawaguchi, Jerzy Golik, George Dubay, Gary Groenewold, Bala Krishnan, Terrence W. Doyle: Esperamicins, a novel class of potent antitumor antibiotics. I. Physico-chemical data and partial structure. In: The Journal of Antibiotics. Band 38, Nr. 11, 1985, S. 1605–1609, doi:10.7164/antibiotics.38.1605.
- ↑ Masataka Konishi, Hiroaki Ohkuma, Kiyoshi Matsumoto, Takashi Tsuno, Hideo Kamei, Takeo Miyaki, Toshikazu Oki, Hiroshi Kawaguchi, Gregory D. Vanduyne, Jon Clardy: Dynemicin A, a novel antibiotic with the anthraquinone and 1,5-diyn-3-ene subunit. In: The Journal of Antibiotics. Band 42, Nr. 9, 1989, S. 1449–1452, doi:10.7164/antibiotics.42.1449.
- ↑ José Cernicharo, Ana M. Heras, A. G. G. M. Tielens, Juan R. Pardo, Fabrice Herpin, Michel Guélin, L. B. F. M. Waters: [ITAL]Infrared Space Observatory's[/ITAL] Discovery of C[TINF]4[/TINF]H[TINF]2[/TINF], C[TINF]6[/TINF]H[TINF]2[/TINF], and Benzene in CRL 618. In: The Astrophysical Journal. Band 546, Nr. 2, 10. Januar 2001, S. L123–L126, doi:10.1086/318871.
- ↑ F. Shindo, Y. Benilan, J.-C. Guillemin, P. Chaquin, A. Jolly, F. Raulin: Ultraviolet and infrared spectrum of C6H2 revisited and vapor pressure curve in Titan's atmosphere. In: Planetary and Space Science. Band 51, Nr. 1, Januar 2003, S. 9–17, doi:10.1016/S0032-0633(02)00151-4.
- ↑ W. C. Maguire, R. A. Hanel, D. E. Jennings, V. G. Kunde, R. E. Samuelson: C3H8 and C3H4 in Titan's atmosphere. In: Nature. Band 292, Nr. 5825, August 1981, S. 683–686, doi:10.1038/292683a0.
- ↑ Jean-Claude Guillemin, Miloud Bouyahyi, El Hassan Riague: Prebiotic, planetary and interstellar chemistry starting from compounds detected in the interstellar medium. In: Advances in Space Research. Band 33, Nr. 1, Januar 2004, S. 81–87, doi:10.1016/j.asr.2003.07.015.
- ↑ The International Union of Pure and Applied Chemistry (IUPAC): acetylenes. In: iupac.org. goldbook.iupac.org, abgerufen am 21. Dezember 2023.
- ↑ a b The International Union of Pure and Applied Chemistry (IUPAC): alkynes. In: iupac.org. goldbook.iupac.org, abgerufen am 21. Dezember 2023.
- ↑ G. P. Moss, P. A. S. Smith, D. Tavernier: Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). In: Pure and Applied Chemistry. Band 67, Nr. 8-9, 1. Januar 1995, S. 1307–1375, doi:10.1351/pac199567081307.
- ↑ Angelika Fallert-Müller, Birgit Jarosch, Angela Simeon: Organische Chemie I. In: Pocket Guide Chemie. Springer Berlin Heidelberg, Berlin, Heidelberg 2019, ISBN 978-3-662-58746-1, S. 127–166, doi:10.1007/978-3-662-58747-8_4.
- ↑ Alois Haas, Hans‐Udo Krächter: Darstellung und Reaktionen Trifluormethylchalkogenyl‐substituierter Alkine. In: Chemische Berichte. Band 121, Nr. 10, Oktober 1988, S. 1833–1840, doi:10.1002/cber.19881211023.
- ↑ Arthur J. Hill, Floyd Tyson: STUDIES ON THE PREPARATION OF THE HIGHER ACETYLENES. I. (PRELIMINARY PAPER.) DEHALOGENATION OF 1,1-DICHLOROHEPTANE IN THE VAPOR PHASE. In: Journal of the American Chemical Society. Band 50, Nr. 1, Januar 1928, S. 172–177, doi:10.1021/ja01388a024.
- ↑ Tsutomu Maruyama, Bunsuke Suzuki: Unsaturated Fatty Acids and Their Derivatives: Part IV. The Mechanism of Forming Stearolic Acid from Dichlorstearic Acid. In: Proceedings of the Imperial Academy. Band 7, Nr. 7, 1931, S. 265–268, doi:10.2183/pjab1912.7.265.
- ↑ N. A. Khan, F. E. Deatherage, J. B. Brown: The preparation of stearolic acid and methyl dideutero-oleate, and certain of their derivatives. In: Journal of the American Oil Chemists Society. Band 28, Nr. 1, Januar 1951, S. 27–31, doi:10.1007/BF02639746.
- ↑ N. A. Khan: Acetylenic compounds. I. The dehydrohalogenation reactions by sodamide in liquid ammonia and preparation of some mono-acetylenic substances and their derivatives. In: Journal of the American Oil Chemists Society. Band 30, Nr. 9, September 1953, S. 355–358, doi:10.1007/BF02633768.
- ↑ John R. Johnson, W. L. McEwen: THE IDENTIFICATION OF MONOSUBSTITUTED ACETYLENES. DERIVATIVES OF DIETHINYL MERCURY. In: Journal of the American Chemical Society. Band 48, Nr. 2, Februar 1926, S. 469–476, doi:10.1021/ja01413a025.
- ↑ a b c d e f Damien Habrant, Vesa Rauhala, Ari M. P. Koskinen: Conversion of carbonyl compounds to alkynes: general overview and recent developments. In: Chemical Society Reviews. Band 39, Nr. 6, 2010, S. 2007, doi:10.1039/b915418c.
- ↑ E.J. Corey, P.L. Fuchs: A synthetic method for formyl→ethynyl conversion (RCHO→RC≡CH or RC≡CR′). In: Tetrahedron Letters. Band 13, Nr. 36, 1972, S. 3769–3772, doi:10.1016/S0040-4039(01)94157-7.
- ↑ a b c d e Frédéric Eymery, Bogdan Iorga, Philippe Savignac: The Usefulness of Phosphorus Compounds in Alkyne Synthesis. In: Synthesis. Band 2000, Nr. 02, 2000, S. 185–213, doi:10.1055/s-2000-6241.
- ↑ Patrick Michel, Dominique Gennet, André Rassat: A one-pot procedure for the synthesis of alkynes and bromoalkynes from aldehydes. In: Tetrahedron Letters. Band 40, Nr. 49, Dezember 1999, S. 8575–8578, doi:10.1016/S0040-4039(99)01830-4.
- ↑ Ernest W. Colvin, Brendan J. Hamill: One-step conversion of carbonyl compounds into acetylenes. In: Journal of the Chemical Society, Chemical Communications. Nr. 5, 1973, S. 151, doi:10.1039/c39730000151.
- ↑ Kazuhiro Miwa, Toyohiko Aoyama, Takayuki Shioiri: Extension of the Colvin Rearrangement Using Trimethylsilyldiazomethane. A New Synthesis of Alkynes. In: Synlett. Band 1994, Nr. 02, 1994, S. 107–108, doi:10.1055/s-1994-22755.
- ↑ Douglass F. Taber, Sha Bai, Peng-fei Guo: A convenient reagent for aldehyde to alkyne homologation. In: Tetrahedron Letters. Band 49, Nr. 48, November 2008, S. 6904–6906, doi:10.1016/j.tetlet.2008.09.114, PMID 19946355, PMC 2634292 (freier Volltext).
- ↑ Y. Taguchi, H. Endo, Y. Abe, J. Matsumoto, T. Wakabayashi, T. Kodama, Y. Achiba, H. Shiromaru: Polyyne formation by graphite laser ablation in argon and propane mixed gases. In: Carbon. Band 94, November 2015, S. 124–128, doi:10.1016/j.carbon.2015.06.058.
- ↑ Seung Keun Shin, Jae Kyu Song, Seung Min Park: Preparation of polyynes by laser ablation of graphite in aqueous media. In: Applied Surface Science. Band 257, Nr. 12, April 2011, S. 5156–5158, doi:10.1016/j.apsusc.2010.10.074.
- ↑ Masaharu Tsuji, Takeshi Tsuji, Shingo Kuboyama, Seong-Ho Yoon, Yozo Korai, Teppei Tsujimoto, Kanji Kubo, Akira Mori, Isao Mochida: Formation of hydrogen-capped polyynes by laser ablation of graphite particles suspended in solution. In: Chemical Physics Letters. Band 355, Nr. 1-2, März 2002, S. 101–108, doi:10.1016/S0009-2614(02)00192-6.
- ↑ Franco Cataldo: Polyynes and cyanopolyynes synthesis from the submerged electric arc: about the role played by the electrodes and solvents in polyynes formation. In: Tetrahedron. Band 60, Nr. 19, Mai 2004, S. 4265–4274, doi:10.1016/j.tet.2004.03.033.
- ↑ a b c d e Alois Fürstner, Paul W. Davies: Alkyne metathesis. In: Chemical Communications. Nr. 18, 2005, S. 2307, doi:10.1039/b419143a.
- ↑ a b Tadashi Eguchi, Kenji Arakawa, Katsumi Kakinuma: Synthesis of over 30-Membered Giant Ring Compounds. Synthetic Study of Archaeal 36- and 72-Membered Macrocyclic Membrane Lipids. In: Journal of Synthetic Organic Chemistry, Japan. Band 57, Nr. 9, 1999, S. 784–797, doi:10.5059/yukigoseikyokaishi.57.784.
- ↑ Rolf Gleiter: Cycloalkadiynes—From Bent Triple Bonds to Strained Cage Compounds. In: Angewandte Chemie International Edition in English. Band 31, Nr. 1, Januar 1992, S. 27–44, doi:10.1002/anie.199200271.
- ↑ Robert A. Benkeser, Gene Schroll, Dale M. Sauve: Reduction of Organic Compounds by Lithium in Low Molecular Weight Amines. II. Stereochemistry. Chemical Reduction of an Isolated Non-terminal Double Bond. In: Journal of the American Chemical Society. Band 77, Nr. 12, Juni 1955, S. 3378–3379, doi:10.1021/ja01617a066.
- ↑ Herbert O. House, Edith F. Kinloch: Reactions involving electron transfer. V. Reduction on nonconjugated acetylenes. In: The Journal of Organic Chemistry. Band 39, Nr. 6, März 1974, S. 747–755, doi:10.1021/jo00920a002.
- ↑ a b Paul J. Kropp, Scott D. Crawford: Surface-Mediated Reactions. 4. Hydrohalogenation of Alkynes. In: The Journal of Organic Chemistry. Band 59, Nr. 11, Juni 1994, S. 3102–3112, doi:10.1021/jo00090a031.
- ↑ a b c d e f g Francis A. Carey, Richard J. Sundberg: Advanced organic chemistry. 5th ed Auflage. Springer, New York 2007, ISBN 978-0-387-44897-8.
- ↑ Robert C. Fahey, Do-Jae Lee: Polar additions to olefins and acetylenes. V. Bimolecular and termolecular mechanisms in the hydrochlorination of acetylenes. In: Journal of the American Chemical Society. Band 90, Nr. 8, April 1968, S. 2124–2131, doi:10.1021/ja01010a034.
- ↑ a b c Suzanne R. Macaulay: The rearrangement of isomeric linear decyn-1-ols by reaction with the sodium salt of 1,3-diaminopropane. In: Canadian Journal of Chemistry. Band 58, Nr. 23, 1. Dezember 1980, S. 2567–2572, doi:10.1139/v80-409.
- ↑ a b Charles Allan Brown, Ayako Yamashita: Saline hydrides and superbases in organic reactions. IX. Acetylene zipper. Exceptionally facile contrathermodynamic multipositional isomerization of alkynes with potassium 3-aminopropylamide. In: Journal of the American Chemical Society. Band 97, Nr. 4, Februar 1975, S. 891–892, doi:10.1021/ja00837a034.
- ↑ Kiitiro Utimoto, Michio Tanaka, Mitsumasa Kitai, Hitosi Nozaki: Cyclization of ω-trimethylsilylethynylalkanoyl chlorides. Application of the preparation of large ring ynones and dl- and (R)-muscone. In: Tetrahedron Letters. Band 19, Nr. 26, 1. Januar 1978, S. 2301–2304, doi:10.1016/S0040-4039(01)91520-5.
- ↑ Kenji Mori, Narshinha P. Argade: Pheromone Synthesis, CLXIII. Synthesis of (9Z,25S,26R,43Z)-25,26-Epoxy-9,43-henpentacontadiene and Its Antipode, Components of the Nymph Recognition Pheromone Produced by Nymphs of the CockroachNauphoeta cinerea. In: Liebigs Annalen der Chemie. Band 1994, Nr. 7, 12. Juli 1994, S. 695–700, doi:10.1002/jlac.199419940711.
- ↑ Vitor L. S. Cunha, Xiaofan Liu, Todd L. Lowary, George A. O’Doherty: De Novo Asymmetric Synthesis of Avocadyne, Avocadene, and Avocadane Stereoisomers. In: The Journal of Organic Chemistry. Band 84, Nr. 23, 24. Oktober 2019, S. 15718–15725, doi:10.1021/acs.joc.9b02391.
- ↑ Nabil Tahiri, Peter Fodran, Dhineshkumar Jayaraman, Jeffrey Buter, Martin D. Witte, Tonatiuh A. Ocampo, D. Branch Moody, Ildiko Van Rhijn, Adriaan J. Minnaard: Total Synthesis of a Mycolic Acid from Mycobacterium tuberculosis. In: Angewandte Chemie International Edition. Band 59, Nr. 19, 10. März 2020, S. 7555–7560, doi:10.1002/anie.202000523, PMID 32067294, PMC 7216993 (freier Volltext).
- ↑ Jack E. Baldwin: Rules for ring closure. In: Journal of the Chemical Society, Chemical Communications. Nr. 18, 1976, S. 734, doi:10.1039/c39760000734.
- ↑ Kerry Gilmore, Rana K. Mohamed, Igor V. Alabugin: The Baldwin rules: revised and extended: Baldwin: Revised, Extended. In: Wiley Interdisciplinary Reviews: Computational Molecular Science. Band 6, Nr. 5, September 2016, S. 487–514, doi:10.1002/wcms.1261.
- ↑ Igor V. Alabugin, Kerry Gilmore: Finding the right path: Baldwin “Rules for Ring Closure” and stereoelectronic control of cyclizations. In: Chemical Communications. Band 49, Nr. 96, 2013, S. 11246, doi:10.1039/c3cc43872d.
- ↑ Kerry Gilmore, Igor V. Alabugin: Cyclizations of Alkynes: Revisiting Baldwin’s Rules for Ring Closure. In: Chemical Reviews. Band 111, Nr. 11, 9. November 2011, S. 6513–6556, doi:10.1021/cr200164y.
- ↑ Noriyuki Takanashi, Kenta Suzuki, Mariko Kitajima, Hiromitsu Takayama: Total synthesis of conolidine and apparicine. In: Tetrahedron Letters. Band 57, Nr. 3, Januar 2016, S. 375–378, doi:10.1016/j.tetlet.2015.12.029.
- ↑ a b Karl Kirchner, Maria José Calhorda, Roland Schmid, Luís F. Veiros: Mechanism for the Cyclotrimerization of Alkynes and Related Reactions Catalyzed by CpRuCl. In: Journal of the American Chemical Society. Band 125, Nr. 38, 1. September 2003, S. 11721–11729, doi:10.1021/ja035137e.
- ↑ a b Yoshihiko Yamamoto: Recent Advances in Intramolecular Alkyne Cyclotrimerization and Its Applications. In: Current Organic Chemistry. Band 9, Nr. 6, S. 503–519, doi:10.2174/1385272053544399.
- ↑ Pablo Wessig, Gunnar Müller: The Dehydro-Diels−Alder Reaction. In: Chemical Reviews. Band 108, Nr. 6, 1. Juni 2008, S. 2051–2063, doi:10.1021/cr0783986.
- ↑ a b Wenbo Li, Liejin Zhou, Junliang Zhang: Recent Progress in Dehydro(genative) Diels–Alder Reaction. In: Chemistry – A European Journal. Band 22, Nr. 5, 26. Januar 2016, S. 1558–1571, doi:10.1002/chem.201503571.
- ↑ Alexander Z. Bradley, Richard P. Johnson: Thermolysis of 1,3,8-Nonatriyne: Evidence for Intramolecular [2 + 4] Cycloaromatization to a Benzyne Intermediate. In: Journal of the American Chemical Society. Band 119, Nr. 41, 1. Oktober 1997, S. 9917–9918, doi:10.1021/ja972141f.
- ↑ Ok Ton Dyan, Gennady I. Borodkin, Pavel A. Zaikin: The Diels–Alder Reaction for the Synthesis of Polycyclic Aromatic Compounds. In: European Journal of Organic Chemistry. Band 2019, Nr. 44, 30. November 2019, S. 7271–7306, doi:10.1002/ejoc.201901254.
- ↑ Young Keun Chung: Transition metal alkyne complexes: the Pauson–Khand reaction. In: Coordination Chemistry Reviews. Band 188, Nr. 1, Juli 1999, S. 297–341, doi:10.1016/S0010-8545(99)00032-6.
- ↑ Rodney A. Fernandes, Anupama Kumari, Ramdas S. Pathare: A Decade with Dötz Benzannulation in the Synthesis of Natural Products. In: Synlett. Band 31, Nr. 05, März 2020, S. 403–420, doi:10.1055/s-0039-1690791.
- ↑ a b Janet Wisniewski Grissom, Gamini U. Gunawardena, Detlef Klingberg, Dahai Huang: The chemistry of enediynes, enyne allenes and related compounds. In: Tetrahedron. Band 52, Nr. 19, Mai 1996, S. 6453–6518, doi:10.1016/0040-4020(96)00016-6.
- ↑ a b Matija Gredičak, Ivanka Jerić: Enediyne compounds - new promises in anticancer therapy. In: Acta Pharmaceutica. Band 57, Nr. 2, 1. Juni 2007, S. 133–150, doi:10.2478/v10007-007-0011-y.
- ↑ Richard R. Jones, Robert G. Bergman: p-Benzyne. Generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-benzenediyl structure. In: Journal of the American Chemical Society. Band 94, Nr. 2, Januar 1972, S. 660–661, doi:10.1021/ja00757a071.
- ↑ Patrick W. Musch, Christian Remenyi, Holger Helten, Bernd Engels: On the Regioselectivity of the Cyclization of Enyne−Ketenes: A Computational Investigation and Comparison with the Myers-Saito and Schmittel Reaction. In: Journal of the American Chemical Society. Band 124, Nr. 8, 1. Februar 2002, S. 1823–1828, doi:10.1021/ja017532f.
- ↑ Michael Schmittel, Marc Strittmatter, Susanne Kiau: Switching from the Myers reaction to a new thermal cyclization mode in enyne-allenes. In: Tetrahedron Letters. Band 36, Nr. 28, Juli 1995, S. 4975–4978, doi:10.1016/0040-4039(95)00937-8.
- ↑ a b Shivalinga Kolle, Sanjay Batra: Transformations of alkynes to carboxylic acids and their derivatives via CC bond cleavage. In: Organic & Biomolecular Chemistry. Band 14, Nr. 47, 2016, S. 11048–11060, doi:10.1039/C6OB01912A.
- ↑ Tanveer Mahamadali Shaikh, Fung-E Hong: Iron-Catalyzed Oxidative Cleavage of Olefins and Alkynes to Carboxylic Acids with Aqueous tert-Butyl Hydroperoxide. In: Advanced Synthesis & Catalysis. Band 353, Nr. 9, Juni 2011, S. 1491–1496, doi:10.1002/adsc.201000899.
- ↑ F. Pennella, R. L. Banks, G. C. Bailey: Disproportionation of alkynes. In: Chemical Communications (London). Nr. 23, 1968, S. 1548, doi:10.1039/c19680001548.
- ↑ André Mortreux, Michel Blanchard: Metathesis of alkynes by a molybdenum hexacarbonyl–resorcinol catalyst. In: J. Chem. Soc., Chem. Commun. Nr. 19, 1974, S. 786–787, doi:10.1039/C39740000786.
- ↑ Alois Fürstner, Günter Seidel: Ring closing alkyne metathesis: stereoselective synthesis of civetone. In: Journal of Organometallic Chemistry. Band 606, Nr. 1, Juli 2000, S. 75–78, doi:10.1016/S0022-328X(00)00096-6.
- ↑ Alois Fürstner, Oliver Guth, Antonio Rumbo, Günter Seidel: Ring Closing Alkyne Metathesis. Comparative Investigation of Two Different Catalyst Systems and Application to the Stereoselective Synthesis of Olfactory Lactones, Azamacrolides, and the Macrocyclic Perimeter of the Marine Alkaloid Nakadomarin A. In: Journal of the American Chemical Society. Band 121, Nr. 48, 1. Dezember 1999, S. 11108–11113, doi:10.1021/ja992074k.
- ↑ Miwako Mori: Synthesis of Natural Products and Related Compounds using Enyne Metathesis. In: Advanced Synthesis & Catalysis. Band 349, Nr. 1-2, 8. Januar 2007, S. 121–135, doi:10.1002/adsc.200600484.
- ↑ Miwako Mori, Tomohiro Tomita, Yoichi Kita, Tsuyoshi Kitamura: Synthesis of (+)-anatoxin-a using enyne metathesis. In: Tetrahedron Letters. Band 45, Nr. 22, Mai 2004, S. 4397–4399, doi:10.1016/j.tetlet.2004.03.171.
- ↑ Jehrod B. Brenneman, Rainer Machauer, Stephen F. Martin: Enantioselective synthesis of (+)-anatoxin-a via enyne metathesis. In: Tetrahedron. Band 60, Nr. 34, August 2004, S. 7301–7314, doi:10.1016/j.tet.2004.06.021.
- ↑ a b Yajing Liu, Jacky W. Y. Lam, Ben Zhong Tang: Conjugated polymers developed from alkynes. In: National Science Review. Band 2, Nr. 4, 1. Dezember 2015, S. 493–509, doi:10.1093/nsr/nwv047.
- ↑ Rongrong Hu, Weizhang Li, Ben Zhong Tang: Recent Advances in Alkyne-Based Multicomponent Polymerizations. In: Macromolecular Chemistry and Physics. Band 217, Nr. 2, Januar 2016, S. 213–224, doi:10.1002/macp.201500291.
- ↑ Rongrong Hu, Jacky W. Y. Lam, Min Li, Haiqin Deng, Jie Li, Ben Zhong Tang: Homopolycyclotrimerization of A 4 -type tetrayne: A new approach for the creation of a soluble hyperbranched poly(tetraphenylethene) with multifunctionalities. In: Journal of Polymer Science Part A: Polymer Chemistry. Band 51, Nr. 22, 15. November 2013, S. 4752–4764, doi:10.1002/pola.26897.
- ↑ Anjun Qin, Jacky W. Y. Lam, Ben Zhong Tang: Click polymerization. In: Chemical Society Reviews. Band 39, Nr. 7, 2010, S. 2522, doi:10.1039/b909064a.
- ↑ Hong-kun Li, Jing-zhi Sun, An-jun Qin, Ben Zhong Tang: Azide-alkyne click polymerization: An update. In: Chinese Journal of Polymer Science. Band 30, Nr. 1, Januar 2012, S. 1–15, doi:10.1007/s10118-012-1098-2.
- ↑ James R. Green: Chemistry of Propargyldicobalt Cations Recent Developments in the Nicholas and Related Reactions. In: Current Organic Chemistry. Band 5, Nr. 7, S. 809–826, doi:10.2174/1385272013375247.
- ↑ John R. Grunwell, Michael F. Wempe, Judith Mitchell, Jocelyn R. Grunwell: The transannular rearrangement of 5-cyclodecynone. In: Tetrahedron Letters. Band 34, Nr. 45, November 1993, S. 7163–7166, doi:10.1016/S0040-4039(00)79277-X.
- ↑ a b c d e Rafael Chinchilla, Carmen Nájera: Recent advances in Sonogashira reactions. In: Chemical Society Reviews. Band 40, Nr. 10, 2011, S. 5084, doi:10.1039/c1cs15071e.
- ↑ a b c d e Rafael Chinchilla, Carmen Nájera: The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. In: Chemical Reviews. Band 107, Nr. 3, 1. März 2007, S. 874–922, doi:10.1021/cr050992x.
- ↑ a b c Dan Wang, Shuanhu Gao: Sonogashira coupling in natural product synthesis. In: Org. Chem. Front. Band 1, Nr. 5, 2014, S. 556–566, doi:10.1039/C3QO00086A.
- ↑ Toyonobu Usuki, Haruka Yamada, Takahiro Hayashi, Hiroto Yanuma, Yohei Koseki, Noriyuki Suzuki, Yoshiro Masuyama, Yong Y. Lin: Total synthesis of COPD biomarker desmosine that crosslinks elastin. In: Chemical Communications. Band 48, Nr. 26, 2012, S. 3233, doi:10.1039/c2cc17958j.
- ↑ Belén Vaz, Leticia Otero, Rosana Álvarez, Ángel R. de Lera: Total Synthesis of Enantiopure Pyrrhoxanthin: Alternative Methods for the Stereoselective Preparation of 4-Alkylidenebutenolides. In: Chemistry - A European Journal. Band 19, Nr. 39, 23. September 2013, S. 13065–13074, doi:10.1002/chem.201301873.
- ↑ Glaser coupling. In: Name Reactions. Springer Berlin Heidelberg, Berlin, Heidelberg 2006, ISBN 3-540-30030-9, S. 263–264, doi:10.1007/3-540-30031-7_117.
- ↑ Siqi Zhang, Liang Zhao: A merged copper(I/II) cluster isolated from Glaser coupling. In: Nature Communications. Band 10, Nr. 1, 24. Oktober 2019, doi:10.1038/s41467-019-12889-w, PMID 31649254, PMC 6813345 (freier Volltext).
- ↑ Allan S. Hay: Oxidative Coupling of Acetylenes. II 1. In: The Journal of Organic Chemistry. Band 27, Nr. 9, September 1962, S. 3320–3321, doi:10.1021/jo01056a511.
- ↑ a b Wei Shi, Aiwen Lei: 1,3-Diyne chemistry: synthesis and derivations. In: Tetrahedron Letters. Band 55, Nr. 17, April 2014, S. 2763–2772, doi:10.1016/j.tetlet.2014.03.022.
- ↑ Guoli Mo, Zaimin Tian, Jiping Li, Guohua Wen, Xiaomin Yang: Silver-catalyzed Glaser coupling of alkynes: Silver-catalyzed Glaser coupling of alkynes. In: Applied Organometallic Chemistry. Band 29, Nr. 4, April 2015, S. 231–233, doi:10.1002/aoc.3275.
- ↑ Riccardo F. Carina, Christiane Dietrich-Buchecker, Jean-Pierre Sauvage: Molecular Composite Knots. In: Journal of the American Chemical Society. Band 118, Nr. 38, 1. Januar 1996, S. 9110–9116, doi:10.1021/ja961459p.
- ↑ a b c d e f g Lambert Brandsma: Synthesis of acetylenes, allenes and cumulenes (= Best synthetic methods). Elsevier, Amsterdam 2004, ISBN 0-12-125751-7.
- ↑ Carin C. C. Johansson Seechurn, Matthew O. Kitching, Thomas J. Colacot, Victor Snieckus: Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. In: Angewandte Chemie International Edition. Band 51, Nr. 21, 21. Mai 2012, S. 5062–5085, doi:10.1002/anie.201107017.
- ↑ a b c d John E. Moses, Adam D. Moorhouse: The growing applications of click chemistry. In: Chem. Soc. Rev. Band 36, Nr. 8, 2007, S. 1249–1262, doi:10.1039/B613014N.
- ↑ a b c d Wolfgang H. Binder, Christian Kluger: Azide/Alkyne-“Click” Reactions: Applications in Material Science and Organic Synthesis. In: Current Organic Chemistry. Band 10, Nr. 14, S. 1791–1815, doi:10.2174/138527206778249838.
- ↑ a b c Jean-François Lutz, Zoya Zarafshani: Efficient construction of therapeutics, bioconjugates, biomaterials and bioactive surfaces using azide–alkyne “click” chemistry. In: Advanced Drug Delivery Reviews. Band 60, Nr. 9, Juni 2008, S. 958–970, doi:10.1016/j.addr.2008.02.004.
- ↑ a b Chao-Jun Li: The Development of Catalytic Nucleophilic Additions of Terminal Alkynes in Water. In: Accounts of Chemical Research. Band 43, Nr. 4, 20. April 2010, S. 581–590, doi:10.1021/ar9002587.
- ↑ E. Yu. Shmidt, I. A. Bidusenko, N. I. Protsuk, A. I. Mikhaleva, B. A. Trofimov: Improved synthesis of tertiary propargyl alcohols by the Favorskii reaction of alkyl aryl (hetaryl) ketones with acetylene. In: Russian Journal of Organic Chemistry. Band 49, Nr. 1, Januar 2013, S. 8–11, doi:10.1134/S1070428013010028.
- ↑ Tao Shen, Teng Wang, Chong Qin, Ning Jiao: Silver-Catalyzed Nitrogenation of Alkynes: A Direct Approach to Nitriles through C≡C Bond Cleavage. In: Angewandte Chemie International Edition. Band 52, Nr. 26, 24. Juni 2013, S. 6677–6680, doi:10.1002/anie.201300193.
- ↑ Alexander Ernst, Luca Gobbi, Andrea Vasella: Orthogonally protected dialkynes. In: Tetrahedron Letters. Band 37, Nr. 44, Oktober 1996, S. 7959–7962, doi:10.1016/0040-4039(96)01838-2.
- ↑ Xin Yang, Daisuke Matsuo, Yoshinori Suzuma, Jing-Kun Fang, Feng Xu, Akihiro Orita, Junzo Otera, Shingo Kajiyama, Nagatoshi Koumura, Kohjiro Hara: Ph2P(O) Group for Protection of Terminal Acetylenes. In: Synlett. Band 2011, Nr. 16, Oktober 2011, S. 2402–2406, doi:10.1055/s-0030-1261223.
- ↑ Reinhard Nast: Coordination chemistry of metal alkynyl compounds. In: Coordination Chemistry Reviews. Band 47, Nr. 1-2, November 1982, S. 89–124, doi:10.1016/0010-8545(82)85011-X.
- ↑ Umberto Belluco, Roberta Bertani, Rino A. Michelin, Mirto Mozzon: Platinum—alkynyl and —alkyne complexes: old systems with new chemical and physical perspectives. In: Journal of Organometallic Chemistry. Band 600, Nr. 1-2, April 2000, S. 37–55, doi:10.1016/S0022-328X(00)00089-9.
- ↑ Helmut Werner: Organometallic chemistry of alkenes and alkynes. In: Journal of Organometallic Chemistry. Band 475, Nr. 1-2, Juli 1994, S. 45–55, doi:10.1016/0022-328X(94)84006-7.
- ↑ Zheng Lu, Khalil A. Abboud, W. M. Jones: Platinum(0) complexes of cyclic alkynes and allenes from base-induced dehydrohalogenation of bromocycloalkenes. In: Organometallics. Band 12, Nr. 4, April 1993, S. 1471–1474, doi:10.1021/om00028a079.
- ↑ Martin A. Bennett, Heinz P. Schwemlein: Metall-Komplexe mit kleinen Cycloalkinen und Arinen. In: Angewandte Chemie. Band 101, Nr. 10, Oktober 1989, S. 1349–1373, doi:10.1002/ange.19891011006.
- ↑ a b Stefanie Federle, Stefanie Hergesell, Sebastian Schubert: Die Stoffklassen der organischen Chemie. Springer Berlin Heidelberg, 2017, S. 40 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Ioan-Teodor Trotuş, Tobias Zimmermann, Ferdi Schüth: Catalytic Reactions of Acetylene: A Feedstock for the Chemical Industry Revisited. In: Chemical Reviews. Band 114, Nr. 3, 12. Februar 2014, S. 1761–1782, doi:10.1021/cr400357r.
- ↑ Ahmed E. S. Nosseir, Angelo Cervone, Angelo Pasini: Review of State-of-the-Art Green Monopropellants: For Propulsion Systems Analysts and Designers. In: Aerospace. Band 8, Nr. 1, 15. Januar 2021, S. 20, doi:10.3390/aerospace8010020.
- ↑ S.D.T. Maduwanthi, R.A.U.J. Marapana: Comparison of pigments and some physicochemical properties of banana as affected by ethephon and acetylene induced ripening. In: Biocatalysis and Agricultural Biotechnology. Band 33, Mai 2021, S. 101997, doi:10.1016/j.bcab.2021.101997.
- ↑ S. D. T. Maduwanthi, R. A. U. J. Marapana: Induced Ripening Agents and Their Effect on Fruit Quality of Banana. In: International Journal of Food Science. Band 2019, 2. Mai 2019, S. 1–8, doi:10.1155/2019/2520179, PMID 31187037, PMC 6521425 (freier Volltext).
- ↑ Frank Z. Stanczyk, David F. Archer, Bhagu R. Bhavnani: Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. In: Contraception. Band 87, Nr. 6, Juni 2013, S. 706–727, doi:10.1016/j.contraception.2012.12.011.
- ↑ Dennis R. Doose, Shean-Sheng Wang, Mukund Padmanabhan, Stefan Schwabe, David Jacobs, Meir Bialer: Effect of Topiramate or Carbamazepine on the Pharmacokinetics of an Oral Contraceptive Containing Norethindrone and Ethinyl Estradiol in Healthy Obese and Nonobese Female Subjects. In: Epilepsia. Band 44, Nr. 4, 10. April 2003, S. 540–549, doi:10.1046/j.1528-1157.2003.55602.x.
- ↑ Frank Z. Stanczyk, David F. Archer: Gestodene: A review of its pharmacology, potency and tolerability in combined contraceptive preparations. In: Contraception. Band 89, Nr. 4, April 2014, S. 242–252, doi:10.1016/j.contraception.2013.12.003.
- ↑ Antony Stewart, Carole Cummins, Lisa Gold, Rachel Jordan, Wendy Phillips: The effectiveness of the levonorgestrel-releasing intrauterine system in menorrhagia: a systematic review. In: British Journal of Obstetrics and Gynaecology. Band 108, Nr. 1, Januar 2001, S. 74–86, doi:10.1016/S0306-5456(00)00020-6.
- ↑ Kusum V. Moray, Himanshu Chaurasia, Oshima Sachin, Beena Joshi: A systematic review on clinical effectiveness, side-effect profile and meta-analysis on continuation rate of etonogestrel contraceptive implant. In: Reproductive Health. Band 18, Nr. 1, Dezember 2021, doi:10.1186/s12978-020-01054-y, PMID 33407632, PMC 7788930 (freier Volltext).
- ↑ M Babják, G Balogh, M Gazdag, S Görög: Estimation of impurity profiles of drugs and related materials. In: Journal of Pharmaceutical and Biomedical Analysis. Band 29, Nr. 6, August 2002, S. 1153–1157, doi:10.1016/S0731-7085(02)00165-6.
- ↑ Susan S. Jick, James A. Kaye, Stefan Russmann, Hershel Jick: Risk of nonfatal venous thromboembolism in women using a contraceptive transdermal patch and oral contraceptives containing norgestimate and 35 μg of ethinyl estradiol. In: Contraception. Band 73, Nr. 3, März 2006, S. 223–228, doi:10.1016/j.contraception.2006.01.001.
- ↑ Karen L Goa, Gregory T Warner, Stephanie E Easthope: Transdermal Ethinylestradiol/Norelgestromin: A Review of its Use in Hormonal Contraception. In: Treatments in Endocrinology. Band 2, Nr. 3, 2003, S. 191–206, doi:10.2165/00024677-200302030-00005.
- ↑ Eric A. Schaff: Mifepristone: ten years later. In: Contraception. Band 81, Nr. 1, Januar 2010, S. 1–7, doi:10.1016/j.contraception.2009.08.004.
- ↑ Herbert Kuhl: Pharmacology of estrogens and progestogens: influence of different routes of administration. In: Climacteric. 2005, Band 8, Nummer sup1, S. 3–63. doi:10.1080/13697130500148875.
- ↑ Richard Godin, Violaine Marcoux: Vaginally Administered Danazol: An Overlooked Option in the Treatment of Rectovaginal Endometriosis? In: Journal of Obstetrics and Gynaecology Canada. Band 37, Nr. 12, Dezember 2015, S. 1098–1103, doi:10.1016/S1701-2163(16)30075-5.
- ↑ Tamás Tábi, László Vécsei, Moussa B. Youdim, Peter Riederer, Éva Szökő: Selegiline: a molecule with innovative potential. In: Journal of Neural Transmission. Band 127, Nr. 5, Mai 2020, S. 831–842, doi:10.1007/s00702-019-02082-0, PMID 31562557, PMC 7242272 (freier Volltext).
- ↑ Vicki Oldfield, Gillian M Keating, Caroline M Perry: Rasagiline: A Review of its Use in the Management of Parkinson??s Disease. In: Drugs. Band 67, Nr. 12, 2007, S. 1725–1747, doi:10.2165/00003495-200767120-00006.
- ↑ Rok Borštnar, Matej Repič, Mojca Kržan, Janez Mavri, Robert Vianello: Irreversible Inhibition of Monoamine Oxidase B by the Antiparkinsonian Medicines Rasagiline and Selegiline: A Computational Study. In: European Journal of Organic Chemistry. 2011, Band 2011, Nummer 32, S. 6419–6433 doi:10.1002/ejoc.201100873.
- ↑ G. M. Everett, L. E. Blockus, I. M. Shepperd: Tremor Induced by Tremorine and Its Antagonism by Anti-Parkinson Drugs. In: Science. Band 124, Nr. 3211, 1956, S. 79–79, doi:10.1126/science.124.3211.79.a.
- ↑ Jin H. Park, Yingting Liu, Mark A. Lemmon, Ravi Radhakrishnan: Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. In: Biochemical Journal. Band 448, Nr. 3, 15. Dezember 2012, S. 417–423, doi:10.1042/BJ20121513, PMID 23101586, PMC 3507260 (freier Volltext).
- ↑ Jonathan Dowell, John D. Minna, Peter Kirkpatrick: Erlotinib hydrochloride. In: Nature Reviews Drug Discovery. Band 4, Nr. 1, Januar 2005, S. 13–14, doi:10.1038/nrd1612.
- ↑ N. Spellmon, C. Li, Z. Yang: Allosterically targeting EGFR drug-resistance gatekeeper mutations. In: Journal of thoracic disease. Band 9, Nummer 7, Juli 2017, S. 1756–1758, doi:10.21037/jtd.2017.06.43, PMID 28839955, PMC 5542946 (freier Volltext).
- ↑ Xiaohai Li, Theodore M. Kamenecka, Michael D. Cameron: Cytochrome P450-Mediated Bioactivation of the Epidermal Growth Factor Receptor Inhibitor Erlotinib to a Reactive Electrophile. In: Drug Metabolism and Disposition. Band 38, Nr. 7, Juli 2010, S. 1238–1245, doi:10.1124/dmd.109.030361, PMID 20382753, PMC 3202369 (freier Volltext).
- ↑ Yahiya Y. Syed: Futibatinib: First Approval. In: Drugs. Band 82, Nr. 18, Dezember 2022, S. 1737–1743, doi:10.1007/s40265-022-01806-z.
- ↑ a b c d Juliana T. W. Tong, Paul W. R. Harris, Margaret A. Brimble, Iman Kavianinia: An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. In: Molecules. Band 26, Nr. 19, 27. September 2021, S. 5847, doi:10.3390/molecules26195847, PMID 34641391, PMC 8510272 (freier Volltext).
- ↑ Yvette N. Lamb: Inotuzumab Ozogamicin: First Global Approval. In: Drugs. Band 77, Nr. 14, September 2017, S. 1603–1610, doi:10.1007/s40265-017-0802-5.
- ↑ Alejandro D. Ricart: Antibody-Drug Conjugates of Calicheamicin Derivative: Gemtuzumab Ozogamicin and Inotuzumab Ozogamicin. In: Clinical Cancer Research. Band 17, Nr. 20, 15. Oktober 2011, S. 6417–6427, doi:10.1158/1078-0432.CCR-11-0486.
- ↑ Gary S. Wood, Jianqiang Wu: Methotrexate and Pralatrexate. In: Dermatologic Clinics. Band 33, Nr. 4, Oktober 2015, S. 747–755, doi:10.1016/j.det.2015.05.009, PMID 26433846, PMC 5549926 (freier Volltext).
- ↑ Lyn C. Guenther: Optimizing Treatment with Topical Tazarotene:. In: American Journal of Clinical Dermatology. Band 4, Nr. 3, 2003, S. 197–202, doi:10.2165/00128071-200304030-00006.
- ↑ Saskia ME Vrouenraets, Ferdinand WNM Wit, Jacqueline van Tongeren, Joep MA Lange: Efavirenz: a review. In: Expert Opinion on Pharmacotherapy. Band 8, Nr. 6, April 2007, S. 851–871, doi:10.1517/14656566.8.6.851.
- ↑ Julia Paik: Lenacapavir: First Approval. In: Drugs. Band 82, Nr. 14, September 2022, S. 1499–1504, doi:10.1007/s40265-022-01786-0, PMID 36272024, PMC 10267266 (freier Volltext).
- ↑ Michele Baldrighi, Davide Bartesaghi, Gabriella Cavallo, Michele R. Chierotti, Roberto Gobetto, Pierangelo Metrangolo, Tullio Pilati, Giuseppe Resnati, Giancarlo Terraneo: Polymorphs and co-crystals of haloprogin: an antifungal agent. In: CrystEngComm. Band 16, Nr. 26, 2014, S. 5897–5904, doi:10.1039/C4CE00367E.
- ↑ S Krishnan-Natesan: Terbinafine: a pharmacological and clinical review. In: Expert Opinion on Pharmacotherapy. Band 10, Nr. 16, November 2009, S. 2723–2733, doi:10.1517/14656560903307462.
- ↑ Kelly Jirschele, Peter K. Sand: Oxybutynin: past, present, and future. In: International Urogynecology Journal. Band 24, Nr. 4, April 2013, S. 595–604, doi:10.1007/s00192-012-1915-8.
- ↑ H. Olschewski: Inhaled iloprost for the treatment of pulmonary hypertension. In: European Respiratory Review. Band 18, Nr. 111, 1. März 2009, S. 29–34, doi:10.1183/09059180.00011111.
- ↑ Marzieh Hashemi, Avat (Arman) Taherpour: Theoretical kinetic and thermodynamic studies of the strain energies and ring size effects of the 1,3-dipolar cycloaddition reactions on ethinamate medicine analogs. In: Journal of Molecular Structure. Band 1204, März 2020, S. 127544, doi:10.1016/j.molstruc.2019.127544.
- ↑ Ulrike Graefe-Mody, Silke Retlich, Christian Friedrich: Clinical Pharmacokinetics and Pharmacodynamics of Linagliptin:. In: Clinical Pharmacokinetics. Band 51, Nr. 7, Juli 2012, S. 411–427, doi:10.2165/11630900-000000000-00000.
- ↑ a b c d e Clemens Lamberth: Alkyne chemistry in crop protection. In: Bioorganic & Medicinal Chemistry. Band 17, Nr. 12, Juni 2009, S. 4047–4063, doi:10.1016/j.bmc.2008.11.037.
- ↑ René Scalla, Michel Matringe, Jean-Michel Camadro, Pierre Labbe: Recent Advances in the Mode of Action of Diphenyl Ethers and Related Herbicides. In: Zeitschrift für Naturforschung C. Band 45, Nr. 5, 1. Mai 1990, S. 503–511, doi:10.1515/znc-1990-0535.
- ↑ Stephen O. Duke, José M. Becerril, T. D. Sherman, John Lydon, Hiroshi Matsumoto: The role of protoporphyrin IX in the mechanism of action of diphenyl ether herbicides. In: Pesticide Science. Band 30, Nr. 4, 1990, S. 367–378, doi:10.1002/ps.2780300402.
- ↑ Brighton Crop Protection Conference 1993 — Weeds: Introduction. In: Crop Protection. Band 12, Nr. 7, November 1993, S. 555–559, doi:10.1016/0261-2194(93)90101-n.
- ↑ L. C. Whelan, M. F. Ryan: Effects of the polyacetylene capillin on human tumour cell lines. In: Anticancer Research. Band 24, Nr. 4, 2004, S. 2281–2286, PMID 15330173.
- ↑ Clemens Lamberth, Andre Jeanguenat, Fredrik Cederbaum, Alain De Mesmaeker, Martin Zeller, Hans-Joachim Kempf, Ronald Zeun: Multicomponent reactions in fungicide research: The discovery of mandipropamid. In: Bioorganic & Medicinal Chemistry. Band 16, Nr. 3, 1. Februar 2008, S. 1531–1545, doi:10.1016/j.bmc.2007.10.019.
- ↑ Yoshio Katsuda, Tadayoshi Chikamoto, Hiroshi Ogami, Hajime Hirobe, Tsutomu Kunishige: Novel Insecticidal Chrysanthemic Esters. In: Agricultural and Biological Chemistry. Band 33, Nr. 9, 1969, S. 1361–1362, doi:10.1271/bbb1961.33.1361.
Verweise
[Bearbeiten | Quelltext bearbeiten]- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Biformin: CAS-Nr.: 1403-08-3, PubChem: 121452, Wikidata: Q76010284.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Siccayne: CAS-Nr.: 22944-03-2, PubChem: 189063, Wikidata: Q82923031.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Onchidin: CAS-Nr.: 161779-98-2, PubChem: 101752790, Wikidata: Q104937330.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Pent-1-en-4-in: CAS-Nr.: 871-28-3, PubChem: 522708, ChemSpider: 455959, Wikidata: Q122577007.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Pyrrhoxanthin: CAS-Nr.: nicht vergeben, PubChem: 16061186, Wikidata: Q76507231.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu 11-Azidoundecanthiol: CAS-Nr.: 668420-70-0, PubChem: 71311045, Wikidata: Q82694993.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Ethinylferrocen: CAS-Nr.: 1271-47-2, PubChem: 139251409, Wikidata: Q72477782.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Norgestimat: CAS-Nr.: 35189-28-7, EG-Nr.: 639-446-1, ECHA-InfoCard: 100.167.085, PubChem: 6540478, ChemSpider: 5022837, DrugBank: DBDB00957, Wikidata: Q7051181.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Norelgestromin: CAS-Nr.: 53016-31-2, EG-Nr.: 642-893-5, ECHA-InfoCard: 100.170.714, PubChem: 62930, ChemSpider: 7843336, DrugBank: DBDB06713, Wikidata: Q15295638.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Tremorin: CAS-Nr.: 1448169-71-8, PubChem: 71621331, DrugBank: DBDB15149, Wikidata: Q76791462.