Sulfate

Sulfate sind eine Gruppe chemischer Verbindungen, die sich von der Schwefelsäure ableiten. Aufteilen lässt sich die Gruppe in Schwefelsäure-Salze und Schwefelsäure-Ester. Die Schwefelsäure besitzt zwei Hydroxy-Gruppen (OH-Gruppen), deren Wasserstoff-Atome abgespalten oder getauscht werden können. Durch Abspaltung dieser H-Atome entstehen aus der Schwefelsäure negativ geladene Anionen: Sulfat [SO4]2− oder Hydrogensulfat [HSO4]−. Diese bilden zusammen mit einem positiv geladenen Gegenion (zum Beispiel Natrium) die Schwefelsäure-Salze, beispielsweise Natriumsulfat (Na2SO4). Bei den Schwefelsäure-Estern sind stattdessen die H-Atome gegen organische Gruppen ausgetauscht. Bei den Monoestern ist ein H-Atom ausgetauscht, bei den Diestern beide.
Die Schwefelsäure ist eine starke Säure, sodass sie in Wasser gelöst überwiegend deprotoniert vorliegt, das heißt als Hydrogensulfat-Ionen. Durch die Abgabe der Wasserstoff-Ionen (H+) wird die Lösung sauer. Schwefelsäure und Hydrogensulfat entstehen in der Atmosphäre durch industrielle Abgase und Vulkanausbrüche und bilden sogenannte Sulfat-Aerosole. Diese sind mitverantwortlich für sauren Regen. Sulfat-Ionen sind in der Umwelt verbreitet, so sind sie nach Chlorid-Ionen die zweithäufigsten Anionen im Meerwasser. Auch in Mineralwasser, in Lebensmitteln und in Salzseen kommen sie vor. Sulfat-Ionen bilden mit Metallen Salze, die zum Teil natürlich als Minerale vorkommen. Das häufigste Salz der Schwefelsäure ist das Calciumsulfat. Es tritt natürlich in mehreren Formen auf, unter anderem als Gips CaSO4·2H2O mit zwei Molekülen Kristallwasser. Ein weiteres häufig vorkommendes Salz ist das Bariumsulfat, das natürlich als Baryt (Schwerspat) vorkommt.
Schwefelsäure-Salze, die nicht (oder nicht reichlich) in der Natur vorkommen, werden industriell hergestellt, wobei in den meisten Reaktionen Schwefelsäure verwendet wird. Diese kann direkt mit einem Metall umgesetzt werden, zum Beispiel zur Herstellung von Chromsulfat aus Chrom. Anderseits können durch die Schwefelsäure auch Sulfate aus Salzen mit anderen Anionen hergestellt werden, beispielsweise Kupfersulfat aus Kupferoxid. Sulfat-Salze sind Zwischenprodukte in der chemischen Industrie, aus ihnen werden oft andere Verbindungen hergestellt. Weiterhin werden sie in der Bauindustrie, als Dünger, als Gerbstoff sowie medizinisch eingesetzt.
Schwefelsäure-Ester kommen als Intermediate im menschlichen und tierischen Metabolismus vor. Verschiedene Giftstoffe wie das Phenol werden im Körper in Sulfate umgewandelt, bevor sie ausgeschieden werden. Körpereigene Verbindungen wie Hormone und das Adrenalin werden im Körper zeitweise in Form ihrer Sulfate gespeichert. Eine andere biologisch wichtige Gruppe sind Vielfachzucker mit Sulfat-Gruppen, die unter anderem als Gerinnungshemmer im Blut sowie als Strukturbestandteil von Knorpel vorkommen. Eine Gruppe von Schwefelsäure-Estern aus Pflanzen sind die Glucosinolate, die vor allem in Kreuzblütlern vorkommen. Ihre Abbauprodukte sind für den charakteristischen Geschmack dieser Pflanzen verantwortlich, unter anderem für die Schärfe von Senf und Meerrettich. Daneben kommen in Tieren, Pflanzen und anderen Lebewesen viele weitere Schwefelsäure-Ester vor.
Um Schwefelsäure-Ester herzustellen, wird vielfach Schwefeltrioxid als Reagenz verwendet. Die direkte Reaktion von Schwefelsäure mit Alkoholen ist auch möglich, aber von untergeordneter Bedeutung. Eine Untergruppe der Sulfate wird als Tenside verwendet. Diese Verbindungen tragen eine lange organische Gruppe, während die zweite OH-Gruppe als Anion vorliegt. Ein Beispiel ist das Natriumlaurylsulfat. Der organische Rest dieser Verbindung enthält zwölf Kohlenstoffatome. Das Gegenion für die negative Ladung ist ein Natrium-Ion. Andere Schwefelsäure-Ester werden als Textilfarbstoffe und als Medikamente verwendet.
Geschichte
[Bearbeiten | Quelltext bearbeiten]Unterschiedliche Formen von Calciumsulfat wurden schon in der Bronzezeit als Baustoff verwendet und auch andere Sulfate werden schon mindestens seit der Antike genutzt. Chemisch beschrieben wurden die ersten Salze aus dieser Gruppe allerdings im 17. Jahrhundert. Die Herstellung von Schwefelsäure-Estern, ihre biologische Bedeutung und ihr natürliches Vorkommen sind erst seit dem 19. Jahrhundert bekannt.
.JPG)
Im östlichen Mittelmeerraum und im Nahen Osten war die Verwendung von Gipsmörtel schon vor Jahrtausenden verbreitet. Im alten Ägypten ist die Verwendung im dritten Jahrtausend vor Christus belegt. Antike Stätten, an denen die Verwendung belegt ist, sind der Alte Palast von Aššur und die Ruinen von Amarna. Im Partherreich wurde Gipsmörtel verwendet, um Gewölbe zu bauen. Von der Zeit des Römischen Reichs bis ins 19. Jahrhundert war Kalkmörtel (mit Calciumcarbonat) wesentlich weiter verbreitet, allerdings wurde auch im Mittelalter zum Teil Gipsmörtel verwendet, zum Beispiel in Frankreich.[1] Die Verwendung von Calciumsulfat in Zement wird seit Ende des 19. Jahrhunderts erforscht, seit den 1930er-Jahren wird es verbreitet eingesetzt.[2] In der Bronzezeit wurde Gips-Alabaster im Minoischen Kreta viel für dekorative Bauelemente verwendet. Gegen Ende der Bronzezeit wurde Gips-Alabaster aus kretischen Steinbrüchen auch anderswo verwendet, beispielsweise für Bänke in Mykene. In den Ruinen von Akrotiri auf Santorini wurde es für Bodenfliesen verwendet.[3] Alabaster ist leicht zu bearbeiten und war im Mittelalter und in der Neuzeit ein verbreitetes und beliebtes Material für Skulpturen und Monumente. Er wurde vor allem in Zentralengland, in Nordspanien und den französischen Alpen abgebaut und über weite Strecken gehandelt. Im Jahre 1550 wurden religiöse Skulpturen in England verboten (siehe Reformatorischer Bildersturm) und im großen Stil Alabasterfiguren nach Frankreich gebracht.[4]
Sulfate wurden vielfach historisch als Farbstoffe und Pigmente verwendet. Im Alten Ägypten wurden Calciumsulfat und Jarosit, KFe3[(OH)6(SO4)2], zur Dekoration von Wänden verwendet.[5] Das Pigment Jarosit wurde daneben in Mittelamerika in Gefäßen aus einer Grabstätte in Teotihuacán gefunden.[6] Im Mittelalter und in der Neuzeit war die Nutzung von Eisen-Gallus-Tinte weitverbreitet. Diese wurde aus Pflanzengalle und Eisen(II)-sulfat zubereitet.[7] Alaun (Kaliumaluminiumsulfat) wurde früher in der Lederherstellung (Gerberei) verwendet, möglicherweise schon im Alten Ägypten.[8] Sicher war es in der Antike in Rom und Griechenland bekannt.[9] Die Technik des Gerbens mit Alaun war in der Antike und im Mittelalter weitverbreitet, obwohl der Effekt auf das Leder nicht permanent war, da das Alaun wieder ausgewaschen werden konnte.[8] Während des gesamten Mittelalters war Alaun ein viel produziertes Industrieprodukt. Neben der Verwendung in der Lederverarbeitung wurde es als Beize in der Wollfärberei verwendet. Ab Mitte des 19. Jahrhunderts wurde es nach und nach durch andere Verbindungen verdrängt, insbesondere durch Aluminiumsulfat, sodass es heute kaum noch Bedeutung hat.[9]
Die ersten Schwefelsäure-Salze waren schon im 17. Jahrhundert als solche bekannt. Natriumsulfat, konkret sein Decahydrat, auch als Glaubersalz bezeichnet, wurde um 1625 von Johann Rudolf Glauber beschrieben. Er analysierte Wasser aus einer Heilquelle in der Nähe von Neapel. Dabei entdeckte er ein Salz, Natriumsulfat, das er Sal mirabile nannte. Jahre später fand er heraus, dass Natriumsulfat aus Steinsalz (Natriumchlorid) und Schwefelsäure hergestellt werden kann.[10] Magnesiumsulfat aus einer Mineralquelle in Epsom, England wurde Ende des 17. Jahrhunderts wissenschaftlich beschrieben. Sowohl das Quellwasser als auch das Salz hatten eine medizinische Wirkung und wurden schon damals als Abführmittel und gegen Kopfschmerzen eingesetzt.[11]
Die medizinische Verwendung von Gips in der Behandlung von Knochenbrüchen begann Anfang des 19. Jahrhunderts. Damals wurden hierzu Holzkästen verwendet, die mit gegossenem Gips aufgefüllt wurden. Diese Technik war zu der Zeit in Europa verbreitet, aber unpraktisch, da die Gipskonstruktionen zu schwer waren, um das Krankenbett damit zu verlassen. Mit Gips fixierte Bandagen für Gipsverbände kamen erst Mitte des 19. Jahrhunderts auf und wurden noch längere Zeit in Krankenhäusern frisch hergestellt. Erst in den 1930er-Jahren wurden gebrauchsfertige Gipsbandagen kommerziell verfügbar.[12] Eine weitere medizinische Errungenschaft ist die Verwendung von Magnesiumsulfat zur Behandlung von Krampfbeschwerden im Rahmen der Schwangerschaft (Eklampsie). Zum ersten Mal eingesetzt wurde die Verbindung in diesem Zusammenhang im Jahr 1916. Bis 1930 hatte Magnesiumsulfat andere weniger geeignete Medikamente (beispielsweise Opioide) bei der Behandlung solcher Beschwerden fast vollständig verdrängt und zu einer deutlichen Verringerung der Müttersterblichkeit geführt.[13]


Seit dem Ende des 19. Jahrhunderts wurde eine größere Zahl an Naturstoffen mit Sulfat-Gruppen entdeckt. So wurde schon 1873 das Pflanzengift Atractylosid im Gummi-Spindelkraut entdeckt.[14] Die Umwandlung von Giftstoffen in Schwefelsäure-Ester in Lebewesen (Sulfatierung) wurde kurz darauf entdeckt. 1876 berichtete Eugen Baumann von der Entdeckung einer unbekannten Verbindung aus Urin. Diese hielt er zunächst irrtümlich für eine Sulfonsäure, identifizierte sie aber kurz darauf korrekt als Kaliumphenylsulfat. In seiner weiteren Arbeit stellte er unter anderem fest, dass eingenommenes Phenol von Menschen und Hunden in diese Verbindung umgewandelt wird, dass das Sulfat wesentlich weniger giftig ist als Phenol selbst und dass Brenzcatechin und Indol ebenfalls Sulfate bilden.[15] Zu den früh entdeckten natürlichen Sulfaten gehören die Senfölglycoside beziehungsweise Glucosinolate aus den Kreuzblütlern. Mehrere Vertreter dieser Gruppe wurden zwischen 1897 und 1899 isoliert, darunter das Gluconasturtiin aus der Brunnenkresse und das Sinalbin aus dem Weißen Senf.[16] Eine weitere Gruppe natürlicher Schwefelsäure-Ester sind die von Flavonoiden abgeleiteten, von denen der erste, Persicarin, 1937 aus dem Wasserpfeffer isoliert wurde.[17] Das Heparin, ein natürlich im Blut vorkommendes gerinnungshemmendes Mittel (Antikoagulans), wurde 1916 entdeckt. Seit 1935 wird es medizinisch eingesetzt.[18] Phosphoadenosinphosphosulfat ist eine chemisch aktivierte Form des Sulfat-Ions, die in Lebewesen zur Bildung von Schwefelsäure-Estern dient. Dieses Molekül wurde in den 1950er-Jahren entdeckt und seine Struktur und Biosynthese wurden aufgeklärt.[19]
Eugène-Melchior Péligot und Jean-Baptiste Dumas stellten 1835 zum ersten Mal Dimethylsulfat und Diethylsulfat her, indem sie Methanol beziehungsweise Ethanol mit Schwefelsäure destillierten. Um 1900 begann in Lyon die industrielle Herstellung von Dimethylsulfat aus Schwefeltrioxid und Dimethylether.[20] Im 20. Jahrhundert wurden verschiedene Synthesemethoden zur Herstellung von Schwefelsäure-Estern entwickelt. So wurden in den 1930er- und 1940er-Jahren Methoden zur Veresterung von Alkoholen mit Schwefeltrioxid und seinen Komplexen entwickelt.[21]
Vertreter und Eigenschaften
[Bearbeiten | Quelltext bearbeiten]
Das Hydrogensulfat (HSO4−) und das Sulfat (SO42−) sind die einfach und zweifach deprononierten Anionen der zweiprotonigen Schwefelsäure (H2SO4). Die Salze, die diese Anionen enthalten, lassen sich demnach in Hydrogensulfate (auch primäre Sulfate) und Sulfate (auch sekundäre Sulfate) einteilen. Bei einwertigen Kationen MI gelten die Summenformeln MIHSO4 und MI2SO4.[22] Alaune sind Doppelsalze aus ein- und dreiwertigen Kationen mit der allgemeinen Summenformel MIMIII(SO4)2 · 12 H2O.[22] Als Vitriole werden die Sulfate zweiwertiger Nebengruppen-Metalle (Kupfervitriol, Eisenvitriol etc.) sowie des Magnesiums bezeichnet, die vier bis sieben Moleküle Kristallwasser enthalten.[23] Die Ester der Schwefelsäure, die eine kovalent gebundene Sulfat-Gruppe enthalten, werden ebenso als Sulfate bezeichnet. Dazu gehören einerseits die Diester, bei denen zwei Moleküle eines Alkohols mit der Säure verestert sind (RO-SO2-OR), andererseits auch die Monoester (RO-SO2-OH) und deren Salze (RO-SO3-MI).[22]
Schwefelsäure-Salze
[Bearbeiten | Quelltext bearbeiten]Die meisten Sulfate sind in Wasser löslich. Ausnahmen bilden die wenig oder schwer löslichen Sulfate einiger Erdalkalimetalle, Calciumsulfat, Strontiumsulfat, Bariumsulfat und Radiumsulfat sowie das Blei(II)-sulfat.[22][24] Das Radiumsulfat ist das am schlechtesten lösliche bekannte Sulfat.[25] Bismut(III)-sulfat, Chrom(III)-sulfat und Quecksilber(II)-sulfat sind in reinem Wasser schlecht löslich, lösen sich aber gut in Säuren.[24] In der folgenden Tabelle ist eine Auswahl an Sulfatsalzen mit ihren Eigenschaften angegeben sowie je zwei Beispiele der Hydrogensulfate und der Alaune.
| Salze der Schwefelsäure | |||||
|---|---|---|---|---|---|
| Name | Formel | Farbe | Wasserlöslichkeit | Schmelzpunkt | Quelle |
| Ammoniumsulfat | (NH4)2SO4 | farblos | hoch | 235 °C (Zersetzung) | [26] |
| Natriumsulfat | Na2SO4 | weiß | hoch | 888 °C | [27] |
| Kaliumsulfat | K2SO4 | farblos bis weiß | hoch | 1067 °C | [28] |
| Magnesiumsulfat | MgSO4 | weiß | hoch | 1124 °C (Zersetzung) | [29] |
| Calciumsulfat | CaSO4 | weiß | gering | 1450 °C | [30] |
| Strontiumsulfat | SrSO4 | weiß | sehr gering | 1605 °C | [31] |
| Bariumsulfat | BaSO4 | weiß | praktisch unlöslich | 1600 °C (Zersetzung) | [32] |
| Aluminiumsulfat | Al2(SO4)3 | weiß | hoch | 770 °C (Zersetzung) | [33] |
| Chrom(III)-sulfat | Cr2(SO4)3 | rotviolett | gering | 700 °C (Zersetzung) | [34] |
| Mangansulfat | MnSO4 | weiß | hoch | 700 °C | [35] |
| Eisen(II)-sulfat | FeSO4 | weiß, als Heptahydrat bläulich oder grünlich | hoch | 400 °C (Zersetzung) | [36] |
| Nickelsulfat | NiSO4 | gelb, als Hexahydrat grün oder blau | hoch | 840 °C (Zersetzung) | [37] |
| Kupfersulfat | CuSO4 | weiß | hoch | 560 °C (Zersetzung) | [38] |
| Zinksulfat | ZnSO4 | Weiß | hoch | 680 °C (Zersetzung) | [39] |
| Natriumhydrogensulfat | NaHSO4 | farblos | hoch | 315 °C (Zersetzung) | [40] |
| Kaliumhydrogensulfat | KHSO4 | farblos | hoch | ca. 195-214 °C (Zersetzung) | [41] |
| Kaliumalaun | KAl(SO4)2·12 H2O | farblos | hoch | 92,5 °C | [42] |
| Chromalaun | KCr(SO4)2·12 H2O | dunkelviolett | hoch | 89 °C | [43] |
Schwefelsäure-Ester
[Bearbeiten | Quelltext bearbeiten]
Symmetrische Dialkylsulfate bis einschließlich Dihexylsulfat[S 1] sind wenig flüchtige Flüssigkeiten.[20] Dimethylsulfat hat einen Schmelzpunkt von −32 °C, eine Dichte von 1,33 g/mL und zersetzt sich ab 188 °C.[44] Diethylsulfat hat einen Schmelzpunkt von −25 °C, eine Dichte von 1,18 g/mL und zersetzt sich ab 205 °C.[45] In Wasser sind die Dialkylsulfate schlecht löslich, in aromatischen Kohlenwasserstoffen und polaren organischen Lösungsmitteln gut löslich. Dialkylsulfate sind starke Alkylierungsmittel. Sie sind reaktiver, je kürzer die Alkylreste sind.[46]
Dimethylsulfat, Diethylsulfat und andere Schwefelsäure-Ester mit kurzkettigen Alkylresten sind sehr giftig. Bei der Exposition gegenüber geringen Konzentrationen Dimethylsulfat treten zunächst nur geringe Symptome auf, jedoch kann es nach stundenlanger Verzögerung zu einem Lungenödem kommen. Durch die Eigenschaften als Alkylierungsmittel wirkt es krebserregend und kann unter anderem Nasenkrebs verursachen. Es wirkt zudem durch Methylierung der DNA mutagen. DNA-Basen und damit das Erbgut werden durch Übertragung von Methylgruppen modifiziert, was zu genetischen Defekten führen kann. Durch die langsamere Hydrolyse ist die akute Toxizität von Diethylsulfat geringer als die von Dimethylsulfat, die Symptome sind jedoch ähnlich und können bis zu Lungenödemen reichen.[46]
Abgrenzung zu anderen Stoffgruppen
[Bearbeiten | Quelltext bearbeiten]
Neben den Sulfaten bildet Schwefel eine Reihe weiterer strukturell verwandte Verbindungen und Verbindungsklassen. Die Sulfite sind die Salze und Ester der schwefligen Säure. Bei diesen weist der Schwefel eine niedrigere Oxidationszahl auf und ist nur an drei Sauerstoffatome gebunden.[47] Bei den Sulfonsäuren trägt der Schwefel drei Sauerstoffatome und eine Kohlenstoffkette.[48] Das Gleiche gilt für deren Salze und Ester, die Sulfonate.[49] Ein Schwefelatom, das zwei Sauerstoffatome und zwei Kohlenstoffketten trägt, kommt in den Sulfonen vor.[50] Bei der Amidosulfonsäure trägt das zentrale Schwefelatom drei Sauerstoffatome und ein Stickstoffatom.[51] Ihre Salze und Ester heißen Sulfamate. Thiosulfate weisen ein zentrales Schwefelatom auf, das neben drei Sauerstoffatomen an ein weiteres Schwefelatom gebunden ist.[52] Ein Kondensationsprodukt aus zwei Molekülen Schwefelsäure ist die Dischwefelsäure.[53] Ihre Salze und Ester werden als Disulfate oder Pyrosulfate bezeichnet.[54]
Das Sulfat-Ion und das Hydrogensulfat-Ion
[Bearbeiten | Quelltext bearbeiten]

Die Bindungsverhältnisse im Sulfat-Ion können entweder durch mesomere Grenzstrukturen mit delokalisierten π-Bindungen und zwei negativ geladenen Sauerstoffatomen oder durch Ladungstrennung mit zweifach positiv geladenem Schwefelatom und negativer Ladung an jedem Sauerstoffatom beschrieben werden. Gilbert Lewis veröffentlichte 1916 die Theorie, dass Atome in Verbindungen bevorzugt acht Außenelektronen tragen (Oktettregel), und sah dies beim Sulfat-Ion als gegeben.[55] Verschiedene moderne Berechnungen stützen diese Ansicht: Die S-O-Bindungen weisen einen erheblichen ionischen Charakter auf, das heißt eine Ladungstrennung. Außerdem müssten zusätzliche Bindungen, die über eine Anzahl von vier hinausgehen, durch d-Orbitale ausgebildet werden. Diese spielen aber vermutlich kaum eine Rolle, da Berechnungen zufolge im zeitlichen Mittel nur 0,19 Elektronen im gesamten Molekül in d-Orbitalen vorliegen. Für ein hexavalentes Schwefelatom wären aber vier d-Elektronen nötig. Obwohl heutiges Wissen klar für eine Struktur mit vierbindigem Schwefel spricht, ist die Darstellung mit sechsbindigem Schwefel verbreitet.[56]
In wässriger Lösung verfügen Ionen über eine sogenannte Hydrathülle aus Wassermolekülen, die über elektrostatische Kräfte an das Ion gebunden sind. Beim Sulfat-Ion besteht die Hydrathülle aus 13 Wassermolekülen.[57] Das Sulfat-Ion weist in Lösung eine Tetraeder-Symmetrie auf. Die S-O-Bindungen sind alle gleichwertig und gleich lang.[58] In kristallinen Feststoffen hängt die Struktur von Sulfat-Ionen von der Zusammensetzung des jeweiligen Salzes ab. Die Winkel zwischen zwei S-O-Bindungen schwanken dabei zwischen 95° und 120°, die Bindungslängen der S-O-Bindungen schwanken zwischen 1,4 und 1,8 Å.[59]
Schwefelsäure ist eine zweiprotonige Säure. Zum pKS-Wert der ersten Deprotonierung der Schwefelsäure liegen in der Literatur sehr unterschiedliche Angaben zwischen −2 und −9 vor. Eine theoretische Analyse aus dem Jahr 2018 kommt beispielsweise auf einen Wert zwischen −4,5 und −8,6.[60] Unabhängig vom genauen pKS-Wert handelt es sich eindeutig um eine starke Säure, die in wässriger Lösung praktisch quantitativ deprotoniert vorliegt. Das Hydrogensulfat-Ion reagiert ebenfalls sauer, ist aber keine starke Säure. Seine Dissoziationskonstante K ist 0,0103,[61] was einem pKS-Wert von 1,99 entspricht. Folglich enthält eine wässrige Lösung kaum undissoziierte Schwefelsäure-Moleküle, sondern hauptsächlich Hydrogensulfat-Ionen und kleinere Mengen an Sulfat-Ionen. Schwefelsäure zeigt eine nennenswerte Autoprotolyse, die ausgeprägter ist als bei Wasser: Bei der Schwefelsäure beträgt die Autoprotolyse-Konstante 3,6, bei Wasser 14. Auch in konzentrierter Schwefelsäure liegen demnach Hydrogensulfat-Ionen vor.[62]
Vorkommen
[Bearbeiten | Quelltext bearbeiten]Freie Sulfat-Ionen sind in der Natur weitverbreitet. Salze des Ions treten in Form mehrerer hundert Minerale auf. Schwefelsäure-Ester spielen eine wichtige Rolle in vielen Lebewesen, inklusive Pflanzen, Tieren und Mikroorganismen.
Vorkommen von Sulfat-Ionen
[Bearbeiten | Quelltext bearbeiten]Sulfat ist eines der häufigsten Anionen in Mineralwasser, neben Chlorid und Hydrogencarbonat.[63] Im Meerwasser ist es das zweithäufigste Anion mit einem Gehalt von 2,71 g/kg oder 2700 ppm.[64][65] Chlorid-Ionen kommen mit gut 19 g/kg in deutlich größerer Menge vor. Alle weiteren Anionen, darunter Bromid und Carbonat, kommen mit unter 0,2 g/kg in deutlich kleineren Mengen vor.[65] Das Wasser in Salzseen enthält oft erhebliche Mengen Sulfat, allerdings ist die Ionen-Zusammensetzung solcher Seen sowohl regional als auch saisonal sehr unterschiedlich.[66] Schwefel ein essenzielles Element für Pflanzen und liegt überwiegend in Form von Sulfat vor, zum Teil zu über 90 %.[67] Sulfat-Ionen sind Ausgangsprodukt der Schwefelassimilation bei Pflanzen und Mikroorganismen. Dabei werden Sulfat-Ionen reduziert und überwiegend in Cystein umgewandelt.[68]

Sulfat ist in allen Lebensmitteln enthalten, der Gehalt ist aber von Produkt zu Produkt sehr verschieden. In einer Studie Anfang der 1990er-Jahre wurden die Sulfat-Gehalte in vielen Lebensmitteln ermittelt. Die höchsten Werte wurden in verarbeiteten Lebensmitteln gefunden, insbesondere in getrockneten Äpfeln (49 μmol/g) und getrockneten Aprikosen (30 μmol/g), etwas niedrigere in anderen Trockenfrüchten wie Rosinen (13 μmol/g) und Datteln (11 μmol/g). Der hohe Gehalt ist dabei vermutlich auf die Konservierung durch Schwefeln zurückzuführen, wobei Schwefeldioxid, Sulfit oder Disulfit zugesetzt wird. Neben Sulfat-Ionen, die als Verunreinigung in Sulfit oder Disulfit vorliegen können, kann Sulfat durch Oxidation der zugesetzten Verbindungen entstehen. Hohe Werte über 10 μmol/g wurden anderweitig in Weizenbrot (13 μmol/g bis 15 μmol/g) und Sojamehl (12 μmol/g) und gefunden. Unter den unverarbeiteten Lebensmitteln enthalten Pflanzen der Gattung Brassica (Kohl) besonders viel freies Sulfat und größere Mengen gebundenes Sulfat in Form von Glucosinolaten (siehe unten). Hierzu gehören Brunnenkresse (11 μmol/g) und Brokkoli, Rosenkohl, Rotkohl und Weißkohl (8 μmol/g bis 10 μmol/g). Frisches Obst und Gemüse enthält ansonsten meist wenig Sulfat. Mandeln und Haselnüsse (je 9 μmol/g) sowie andere Nüsse und Samen enthalten jedoch höhere Mengen.[69]
Minerale
[Bearbeiten | Quelltext bearbeiten]Viele Metallsulfate kommen in der Natur in Form von Mineralen vor. Die Sulfatminerale weisen überwiegend ein nichtmetallisches Aussehen auf sowie eine geringe Dichte und Härte.[70] Viele Vertreter der Gruppe sind wasserlöslich und wenig beständig.[71] Die Sulfate der Erdalkalimetalle sind besonders stabil und am weitesten verbreitet.[70] Sulfatminerale können auf unterschiedlichen Wegen gebildet werden. Dazu gehört einerseits die Ausfällung aus wässriger Lösung (sowohl im Meer als auch in Seen) andererseits die Oxidation von Sulfiden und anderen Schwefel-Verbindungen.[71]
In den gängigen Mineralsystematiken bilden die Sulfatminerale eigene Systemklassen. In der von der International Mineralogical Association (IMA) zuletzt 2009 aktualisierten 9. Auflage der Mineralsystematik nach Strunz bilden die Sulfate eine Klasse, zu der auch die Selenate, Tellurate, Chromate, Molybdate und Wolframate gerechnet werden.[72] Die Lapis-Systematik leitet sich von der alten Systematik nach Strunz in der 8. Auflage ab und enthält analog eine Klasse Sulfatminerale (einschließlich Chromate, Molybdate und Wolframate).[73] In der Mineralsystematik nach Dana bilden die Sulfate zusammen mit den Chromaten und Molybdaten eine eigene Klasse, die aus neun Abteilungen besteht. Aufgeteilt sind diese je nach Vorhandensein von Kristallwasser sowie weiteren Anionen.[74][75] Stand 2018 waren rund 450 Sulfat-Minerale bekannt.[73]
Calciumsulfat
[Bearbeiten | Quelltext bearbeiten]Calciumsulfat (CaSO4) ist das mit Abstand am weitesten in der Natur verbreitete Sulfat und tritt in mehreren Mineralarten und -varietäten auf. Mit Kristallwasseranteil wird es als Gips (CaSO4·2H2O) oder Bassanit (CaSO4·½H2O) bezeichnet, ohne als Anhydrit. Gips ist das häufigste Sulfatmineral, weltweit existieren viele Vorkommen und enorme Reserven.[76][77] Es ist ein Evaporit-Mineral, das heißt, es entsteht durch Ausfällung aus wässriger Lösung, wenn diese durch Verdunstung aufkonzentriert wird. Möglich ist dies bei Meerwasser und in Seen, wenn diese eine hohe Sulfatkonzentration aufweisen.[76] Die Minerale Gips, Bassanit und Anhydrit können sich in Abhängigkeit der Feuchtigkeit oder der Trockenheit der Umgebung leicht ineinander umwandeln.[78] Wird Gips unter anderem Material begraben, führen erhöhter Druck und erhöhte Temperatur (ab 42 °C) zur Entwässerung und Bildung von Anhydrit. Gelangt der Anhydrit wieder an die Erdoberfläche, wird mit der Zeit Gips zurückgebildet. Oft treten deshalb beide Formen gemeinsam auf.[79]
-
 Gipskristall
Gipskristall -
 Bassanit
Bassanit -
 Anhydritkristalle
Anhydritkristalle



In einigen Weltregionen liegen geeignete Bedingungen für die Neubildung von Gips vor. Ein Beispiel für die Bildung aus Meerwasser ist der Persische Golf; Seen, an denen Gips gebildet wird, sind zum Beispiel der Große Salzsee in Utah (Vereinigte Staaten), die Laguna de Callocanta (Spanien) und der Tschad-See. Besonders alte Gips-Vorkommen liegen in Australien und Nordwestrussland. Geologisch jüngere Vorkommen existieren in Kanada und den Vereinigten Staaten, in Mitteleuropa (Zechstein) und in den Anden.[76] Im White-Sands-Nationalpark in New Mexico gibt es Sanddünen, die zu 96 % aus feinen Gipskörnern bestehen.[80] Eine reinweiße und extrem feinkörnige Gipsvariante wird Alabaster genannt, transparente Gips-Einkristalle bilden das Marienglas.[81] In der Mine von Naica in Mexiko kommen riesige Kristalle aus Marienglas vor, die bis zu elf Meter lang und einen Meter dick sind.[82] Sandrosen bestehen aus verwachsenen Gipskristallen und Sandkörnern.[83] In der Atacama-Wüste gibt es größere Vorkommen von Calciumsulfat, die aufgrund der Trockenheit hauptsächlich aus Anhydrit mit einer dünnen Gipsschicht bestehen.[78] Vorkommen von Bassanit sind selten, er wurde aber in der kalifornischen Wüste, am Vesuv und in Ölsanden aus Zentralasien nachgewiesen,[84] außerdem in der Cioclovina-Höhle in Rumänien.[85] Anhydrit, Bassanit und Gips wurden auch auf dem Mars entdeckt.[86] In der nördlichen Polarregion des Planeten wurde Calciumsulfat nachgewiesen, bei dem es sich vermutlich überwiegend um Gips handelt.[87]
-
 White-Sands-Nationalpark
White-Sands-Nationalpark -
 Kristalle in der Mine von Naica; unten rechts ist klein eine Person zu sehen
Kristalle in der Mine von Naica; unten rechts ist klein eine Person zu sehen -
 Sandrose
Sandrose



Andere Minerale
[Bearbeiten | Quelltext bearbeiten]

Die bedeutendsten Sulfatminerale neben den Calciumsulfaten sind Baryt (Schwerspat, BaSO4), Coelestin (SrSO4), Anglesit (PbSO4) und Epsomit (MgSO4·7 H2O).[70][88] Anglesit bildet sich als Sekundärmineral aus Galenit (PbS). Oft wird Galenit jedoch stattdessen zu Cerussit (PbCO3) umgewandelt, sodass Anglesit im Gegensatz zu diesen beiden Mineralen seltener ist.[89] Kupfersulfat-Pentahydrat kommt natürlich als Chalkanthit vor.[90] Kaliumsulfat kommt natürlich in Form des Minerals Arcanit vor, das in Guano-Ablagerungen und Fumarolen gefunden wurde.[91] Es ist eine zweistellige Zahl an Sulfat-Mineralen bekannt, die in und um Salzseen vorkommen. Häufige Vertreter neben Gips sind Blödit (Na2Mg[SO4]2·4 H2O), Mirabilit (Na2SO4·10 H2O), Thenardit (Na2SO4) sowie Epsomit (MgSO4·7 H2O) und Hexahydrit (MgSO4·6 H2O).[66]
Biominerale
[Bearbeiten | Quelltext bearbeiten]
Biominerale sind mineralische Verbindungen, die von Lebewesen gebildet werden. Sie dienen unter anderem als Sinnesorgane und als Strukturelemente im Skelett. Verschiedene Sulfat-Minerale kommen als Biominerale in Lebewesen vor, vorwiegend in solchen, die im Meer leben.[92] Die einzelligen Acantharia besitzen ein Skelett, das aus zehn oder zwanzig Skleriten (Skelett-Segmenten) besteht. Bei diesen handelt es sich jeweils um Einkristalle aus Coelestin (Strontiumsulfat).[93] Sulfatminerale, insbesondere Bassanit (CaSO4 · ½ H2O), kommen als Biominerale in sogenannten Statolithen bei Quallen vor. Dabei handelt es sich um kleine Kristalle, die der Wahrnehmung der Schwerkraft dienen.[94] Die Schirmquallen und Würfelquallen besitzen beide Statolithen aus Calciumsulfat.[95] Bei den Statolithen der Kronenqualle handelt es sich um Bassanit-Einkristalle mit einer Länge von etwa 60 μm und einem Durchmesser von etwa 15 μm.[96] Statolithen von Würfelquallen der Gattung Carbybdea bestehen ebenfalls aus Bassanit, allerdings in Form eines Clusters anstatt in Form von Einkristallen.[95] Bassanit wird normalerweise leicht zu Gips hydratisiert, was aber durch Einbettung in Membranen verhindert wird. Mit einer 32 % höheren Dichte gegenüber Gips ist Bassanit zum Zweck der Gravitationswahrnehmung vorteilhaft.[96][97] Sulfat kann in geringen Mengen in Muschelschalen eingebaut werden, die überwiegend aus Aragonit (Calciumcarbonat) bestehen. Bei der Riesenmuschel-Art Hippopus hippopus wurden in der Schale Sulfat-Anteile bis über 0,1 % nachgewiesen.[98] Baryt zur Gravitationswahrnehmung kommt in Algen der Gattung Chara vor.[99] Neben den Vorkommen in Meereslebewesen wurden im Holz des Zahnbürstenbaums Bassanit-Kristalle nachgewiesen.[100]
Atmosphärenchemie der Sulfate
[Bearbeiten | Quelltext bearbeiten]
Sulfat-Aerosole bestehen vorwiegend aus dissoziierter Schwefelsäure, das heißt in Form von gelösten Hydrogensulfat-Ionen. Sie haben einen erheblichen Einfluss auf das Klima und können insbesondere vorübergehende Abkühlungen verursachen.[101][102] Sulfat-Aerosole entstehen aus Schwefeldioxid, das im Wesentlichen aus drei Quellen stammt: Industrielle Abgase, Vulkanausbrüche und Oxidation von Dimethylsulfid, das durch Phytoplankton freigesetzt wird. Durch Oxidation des Schwefeldioxids, beispielsweise durch Hydroxylradikale, und Reaktion mit Wasser wird in der Atmosphäre Schwefelsäure gebildet. Schwefelsäure- und Wassermoleküle bilden feine Partikel, wobei sich durch Dissoziation der Säure hauptsächlich Hydrogensulfat-Ionen bilden. Alternativ kann sich Schwefeldioxid im Wasser vorhandener Wolken lösen, wo es durch Wasserstoffperoxid oxidiert wird. Auch in diesem Fall bildet sich Schwefelsäure, die jedoch sofort in Lösung vorliegt und zu Hydrogensulfat dissoziiert. In beiden Fällen können Schwefelsäure und Hydrogensulfat durch Reaktion mit Ammoniak zum Teil zu Ammoniumsulfat weiterreagieren. Die abkühlende Wirkung der Sulfat-Aerosole beruht einerseits auf der Eigenschaft der Partikel, Sonnenlicht zu reflektieren. Andererseits wirken sie als Kondensationskeime für die Wolkenbildung und erhöhen die Dichte und die Albedo (die Reflexionsfähigkeit) der Wolken.[103]
Sulfat-Aerosole werden oft innerhalb kurzer Zeit ausgewaschen und können sich dadurch nicht in der gesamten Atmosphäre verbreiten. Der Kühlungs-Effekt ist daher überwiegend zeitlich und örtlich begrenzt.[103] Gemäß einer wissenschaftlichen Studie hat die Freisetzung von Schwefeldioxid durch die Industrie in bestimmten Weltregionen vorübergehend die Klimaerwärmung durch Treibhausgase ausgeglichen, insbesondere in Mitteleuropa und den östlichen Vereinigten Staaten.[104] Speziell bei größeren Vulkanausbrüchen wird das freigesetzte Schwefeldioxid bis in die Stratosphäre getragen, wo die Aerosolpartikel mehrere Jahre verweilen können und dann globale Klimaeffekte verursachen. Aus der Analyse von Eisbohrkernen können anhand von Sulfat-Ablagerungen Rückschlüsse auf Vulkanausbrüche in der Vergangenheit gezogen werden.[105] Sulfat-Aerosole tragen zum sauren Regen bei. Das Schwefeldioxid gehört neben Kohlendioxid und Stickoxiden zu den wichtigsten Gasen, die in der Atmosphäre zu Säuren reagieren können. Schwefelsäure (aus Schwefeldioxid) ist eine starke Säure. Sie dissoziiert daher vollständig zu Hydrogensulfat und Oxonium-Ionen. Die Konzentration an Oxonium-Ionen korreliert direkt mit dem pH-Wert, daher sind die Sulfat-Aerosole stark sauer und ihre Auswaschung aus der Atmosphäre führt zu saurem Regen.[106]

Beispiele für Vulkanausbrüche, die zu einer vorübergehenden Abkühlung des Klimas führten, sind der Ausbruch des Krakatau 1883 und der Ausbruch des Pinatubo 1991.[103] Die durch den Ausbruch des Pinatubo 1991 freigesetzte Menge an Schwefeldioxid betrug etwa 20 Millionen Tonnen. Durch die entstehenden Sulfat-Aerosole war die globale Durchschnittstemperatur 1992 um 0,4 °C kälter als erwartet und 1993 um 0,1 °C.[107] Der Ausbruch des Tambora 1815 setzte ungefähr 60 bis 80 Millionen Tonnen Schwefeldioxid frei und führte weltweit und besonders in Europa zu deutlichen klimatischen Abkühlungen in den folgenden Jahren, inklusive des sogenannten Jahrs ohne Sommer 1816.[108] In den Jahren 536 und 540 fanden vermutlich zwei Vulkanausbrüche statt, die etwa 30 und 50 Millionen Tonnen Schwefeldioxid freisetzten. Die dadurch erzeugten Sulfat-Aerosole führten zu einer deutlichen Abschwächung der Sonneneinstrahlung und zu einer vorübergehenden Verringerung der weltweiten durchschnittlichen Temperatur um mehrere Grad Celsius. Das Jahrzehnt ab 536 war das durchschnittlich kälteste in den vergangenen zwei Jahrtausenden (siehe Klimaanomalie 536–550).[109]
Allgemeine biologische Bedeutung
[Bearbeiten | Quelltext bearbeiten]
Sulfat-Ionen sind wenig reaktiv und werden sowohl in der Schwefel-Assimilation als auch für die Biosynthese von Schwefelsäure-Estern aktiviert, meist als Phosphoadenosinphosphosulfat (PAPS). Durch Reaktion von Sulfat mit Adenosintriphosphat (ATP) entsteht das gemischte Anhydrid Adenosinphosphosulfat. Dessen Phosphorylierung ergibt PAPS.[68] Bei Tieren werden beide Syntheseschritte des PAPS vom gleichen Enzym, der PAPS-Synthase, katalysiert. In Pflanzen, Pilzen und Bakterien werden die beiden Schritte jeweils von zwei verschiedenen Enzymen katalysiert.[110]
Die Schwefel-Assimilation ist die Aufnahme von Schwefel-Verbindungen und deren Umwandlung in Verbindungen, die für den jeweiligen Organismus besser verwendbar sind. Bei diesem Prozess wird in Pflanzen und Mikroorganismen zunächst aus Sulfat entweder Adenosinphosphosulfat oder PAPS gebildet. Diese Verbindungen reagieren mit Thiol-Gruppen in Proteinen. Dadurch bilden sich proteingebundene Thiosulfat-Gruppen. Eine solche Gruppe kann durch eine Thiosulfatreduktase zu einer Disulfid-Gruppe reduziert werden. Das zweite Schwefelatom dieser Gruppe wird anschließend unter Abspaltung von Acetat auf O-Acetylserin übertragen, wodurch Cystein entsteht.[68]
Die Biosynthese von Sulfaten und die dafür nötigen Sulfotransferasen kommen bei allen Lebewesen vor. Als Quelle für die Sulfat-Gruppen dient dabei das PAPS. Sulfotransferasen kommen einerseits im Cytosol (in der Zellflüssigkeit) und andererseits membranständig (an die Zellmembran gebunden) vor. Cytosolische Sulfotransferasen setzen vorwiegend kleine Moleküle um. Dazu gehören die Regulierung von Steroidhormonen und die Entgiftung von organismusfremden Stoffen. Die membranständigen Sulfotransferasen modifizieren überwiegend große Biomoleküle wie Proteine und Kohlenhydrate. Die Sulfatierung großer Biomoleküle spielt eine Rolle bei vielen biochemischen Prozessen. Modifikationen von Proteoglycanen steuern die Eigenschaften für die zelluläre Signaltransduktion und Molekülerkennung. Weitere Prozesse, bei denen sulfatierte Makromoleküle beteiligt sind, sind der Eintritt von Viren in Zellen und die Regulierung der Blutgerinnung.[110][111] Wichtig für viele biologische Eigenschaften von Biomolekülen mit Sulfat-Gruppen ist die Tatsache, dass diese Gruppen bei physiologischen pH-Werten grundsätzlich deprotoniert vorliegen, sodass diese Biomoleküle insgesamt eine erhebliche negative Ladung aufweisen. Dies beeinflusst wiederum die Löslichkeit, kann die Konformation der Moleküle ändern und ermöglicht ionische Wechselwirkungen mit anderen Molekülen.[110] Neben den weitverbreiteten Sulfotransferasen, die PAPS als Sulfatquelle nutzen, kommen bei Bakterien auch noch PAPS-unabhängige Sulfotransferasen vor. Diese können Sulfat-Gruppen von einem Phenylester auf ein Phenol übertragen, wobei ein anderer Phenylester entsteht.[112]
Vorkommen in Tieren
[Bearbeiten | Quelltext bearbeiten]In vielzelligen Tieren ist die Sulfatierung chemischer Verbindungen ein wichtiger biologischer Prozess, der in allen Organen stattfindet. Das für die Sulfatierung nötige PAPS kann bei Säugetieren in allen Geweben gebildet werden. Stand 2002 waren 44 cytosolische Sulfotransferasen aus Säugetieren bekannt sowie 32 membranständige Sulfotransferasen.[110] Aus dem Cytosol menschlicher Zellen waren Stand 2004 zehn Sulfotransferasen bekannt.[111]
Sulfatierung im Metabolismus kleiner Moleküle
[Bearbeiten | Quelltext bearbeiten]Teilweise dient die Sulfatierung endogener (körpereigener) Substanzen dazu, bioaktive Verbindungen in eine stabile, inaktive Speicherform zu überführen. Sulfotransferasen, die der Regulierung von Hormonen dienen, sind vergleichsweise selektiv für ihre jeweiligen Substrate. Sulfotransferasen, die der Entgiftung dienen, können demgegenüber eine größere Bandbreite an Molekülen umsetzen. Die Estrogen-Sulfotransferase reguliert das Mengenverhältnis zwischen Estrogenen und ihren Sulfaten.[111] Auch das Hormon Pregnenolon wird reguliert, indem es teilweise in das inaktive Pregnenolonsulfat[S 2] umgewandelt wird. Freie Catecholamine (darunter Dopamin und Adrenalin) werden schnell abgebaut und liegen überwiegend in modifizierter Form vor. Die genauen Modifikationen unterscheiden sich zwischen verschiedenen Säugetier-Arten. Bei Menschen ist die Sulfatierung die häufigste Modifikation, während es bei Ratten die Glucuronidierung ist. Im Gegensatz zu anderen Reaktionen der Catecholamine ist die Sulfatierung reversibel.[110]

Die Sulfatierung ist außerdem eine verbreitete Form der Konjugatbildung. Viele Fremdstoffe, inklusive Medikamenten, werden durch die Bildung von Sulfaten und anderen Konjugaten in eine biologisch weniger aktive Form überführt. Eine Ausnahme stellt der Arzneistoff Minoxidil dar, dessen im Körper gebildetes Sulfat die eigentlich aktive Verbindung ist. Die für die Konjugatbildung nötigen Sulfotransferasen kommen vermehrt in der Leber vor. Bei schlecht wasserlöslichen Verbindungen wird durch die Sulfatierung die Wasserlöslichkeit erhöht und die Ausscheidung erleichtert. Zu den körperfremden Verbindungen, die so modifiziert werden, gehören die Phenole, beispielsweise das para-Nitrophenol.[110][113] Phenol selbst war die erste Verbindung, bei der die Metabolisierung durch Bildung eines Sulfats nachgewiesen wurde.[15] Im Fettsäurestoffwechsel werden die dabei gebildeten Gallensäuren sulfatiert.[114] Über die Nahrung aufgenommene Flavonoide werden ebenso sulfatiert, so wird aus Quercetin und seinen Glycosiden das Quercetin-3'-O-sulfat gebildet.[115] Andere körperfremde Stoffe, die im Metabolismus nachweislich zu Sulfaten umgesetzt werden, sind die Medikamente Salbutamol und Levosalbutamol.[116] Daneben werden auch überschüssige körpereigene Verbindungen über diesen Weg eliminiert. Beispiele hierfür sind die Schilddrüsenhormone Triiodthyronin und Tetraiodthyronin[110] und die Aminosäure Tryptophan, die im Körper zu Indoxylsulfat abgebaut wird.[117]
Sulfatierung von Kohlenhydraten
[Bearbeiten | Quelltext bearbeiten]Glycosaminoglycane sind Kohlenhydrat-Ketten, die aus zwei sich abwechselnden Zucker-Einheiten bestehen: Einer Uronsäure und einem Aminozucker, die zusätzlich eine variable Anzahl an Sulfat-Gruppen aufweisen. Die Verbindungen weisen eine große Strukturvielfalt auf, was Kettenlänge und Sulfatierungsgrad angeht. Bei den sogenannten Proteoglycanen sind Glycosaminoglycan-Ketten an Proteine gebunden. Die Proteoglycane erfüllen vielfältige biologische Funktionen, unter anderem beim Aufbau von Geweben und der biochemischen Signaltransduktion.[110][118] Bei der Biosynthese von Glycosaminoglycanen durch Sulfotransferasen werden zunächst allgemeine Vorläufermoleküle gebildet, die zum Ende der Biosynthese an eine bestimmte biologische Funktion angepasst werden.[111] Die Polysaccharid-Kette wird dabei sulfatiert, wobei im Allgemeinen mehr als eine Sulfat-Gruppe pro Wiederholeinheit auftreten. Das exakte Muster der Sulfat-Gruppen ist aber je nach genauer Funktion der Moleküle unterschiedlich.[118]

Beim Chondroitinsulfat sind die Zucker-Einheiten Glucuronsäure und N-Acetylgalactosamin. Chondroitinsulfat-Varianten regulieren die Vermehrung von Stammzellen und das Wachstum von Nervenzellen (Neurogenese) und sind an der Bildung des Gehirns beteiligt.[118] Chondroitinsulfate sind außerdem Bestandteil des Proteoglycans Aggrecan, das eine Hauptstrukturkomponente von Knorpel ist.[119] Die Hauptkomponenten von Knorpel neben Wasser sind Kollagen und Aggrecan. Durch die negative Ladung der Sulfat-Gruppen am Aggrecan nimmt der Knorpel Wasser auf und schwillt an, dieser Kraft entgegenwirken die Kollagenfasern. Dieses Zusammenspiel ist die Grundlage des charakteristischen elastischen Verhaltens von Knorpel.[120] Chondroitinsulfate kommen in sehr unterschiedlichen Tieren vor, sowohl in Säugetieren (inklusive Menschen) als auch in Wirbellosen wie Tintenfischen.[121] Beim Keratansulfat sind die Einheiten Galactose und N-Acetyllactosamin,[S 3] es ist das einzige der Glycosaminoglycane, das statt einer Uronsäure einen nicht oxidierten Zucker (Galactose) enthält. Eine geringe Anzahl von Keratansulfat-Einheiten kommt neben Chondroitinsulfat im Aggrecan in Knorpel vor. Besonders viel kommen Keratansulfat-Proteoglycane in der Hornhaut vor, wo sie dazu dienen, das Auge feucht und damit die Hornhaut transparent zu halten.[122] Beim Dermatansulfat sind die Zucker-Einheiten Iduronsäure und N-Acetylgalactosamin.
Dermatansulfat kommt primär in der unteren Hautschicht (Dermis) vor, nach der es benannt ist. Es macht bis zu 0,3 % des Trockengewichts der Haut aus. Dermatansulfat ist dort an der Wundheilung beteiligt.[123] Dermatansulfat entsteht aus Chondroitinsulfat: Durch eine Epimerisierungs-Reaktion ändert sich die Stereokonfiguration der Glucuronsäure, wodurch die Iduronsäure-Einheiten entstehen.[124] Beim Heparansulfat sind die Zucker-Einheiten Glucuronsäure oder Iduronsäure und N-Acetylglucosamin.[18] Funktionalisiertes Heparansulfat ist unter anderem wichtig für die Regulierung der Blutgerinnung und die Angiogenese (Bildung von Blutgefäßen).[18][111] Heparin besteht aus denselben Zucker-Einheiten wie das Heparansulfat, allerdings in anderen Mengenverhältnissen, außerdem weist das Heparin mehr Sulfat-Gruppen auf.[18] Es wirkt gerinnungshemmend und dient der körpereigenen Regulierung der Blutgerinnung. Für die Wirkung ist dabei eine spezifische Abfolge von fünf Zucker-Einheiten essenziell, die in der Kette mehrfach vorliegen kann.[18][125] Die Anbindung an Glycosaminoglycane ist für Pathogene teilweise ein wichtiger Mechanismus, um Zellen zu erkennen und anschließend in diese einzudringen. Für den parasitischen Malaria-Erreger Plasmodium falciparum handelt es sich um Chondroitinsulfat, das einen geringen Sulfatierungsgrad aufweisen muss, beim Herpes-Simplex-Virus einen hohen.[118] Das Dermatansulfat-Proteoglycan Decorin ist wichtig als Bindungspunkt für den Borreliose-Erreger Borrelia burgdorferi.[123] Dengue-Viren binden über Heparansulfat an Zellen.[126]
Schleim auf Schleimhäuten enthält als strukturgebende Komponente Mucine. Diese sind Glycoproteine, das heißt kohlenhydrat-modifizierte Proteine. Die Kohlenhydratkomponenten der Mucine bestehen überwiegend aus Fucose, Galactose, N-Acetylgalactosamin und N-Acetylglucosamin sowie Sialinsäuren. Zusätzlich sind sie teilweise mit Sulfat-Gruppen modifiziert.[127] Der Schleim erfüllt auf den Schleimhäuten mehrere Funktionen, darunter als Gleitmittel, zum Erhalt einer Feuchtigkeitsschicht und als Schutzschicht gegen Pathogene und andere schädliche Einflüsse.[128] Mit Wasser bilden die Mucine ein Gel, das für die Konsistenz des Schleims verantwortlich ist. Durch die Sialinsäure- und Sulfat-Gruppen sind die Mucine negativ geladen, wodurch sie Wasser und Kationen wie Calcium binden, die zusätzliche elektrostatische Wechselwirkungen zwischen Molekülen ermöglichen. Die negative Ladung ist demnach ein zentraler Faktor für die Fähigkeit zur Gelbildung.[127]

Glycolipide sind Biomoleküle, bei denen ein Lipid, zum Beispiel ein Glycerinester, zusätzlich ein oder mehr Zucker-Einheiten trägt. Glycolipide mit Sulfat-Gruppen, beispielsweise Sphingolipide, kommen im Myelin, in der Leber und in Spermatozoen vor.[110] In Myelin und Leber kommen die Sulfatide vor. Dabei handelt es sich um Ceramid-Lipide, die zusätzlich eine sulfatierte Galactose-Einheit tragen. In Spermatozoen kommen die Seminolipide vor, die eine ähnliche Struktur mit einer sulfatierten Galactose-Einheit aufweisen. Im Gegensatz zu den Ceramid-Derivaten tragen sie an der Glycerin-Einheit jedoch keine Amid-Gruppe.[129] Im Gehirn und Nervensystem kommen noch weitere strukturell verwandte Verbindungen vor. Sulfatide steuern die Bildung und den Erhalt von Nervenfasern. In Tierversuchen an Mäusen konnte gezeigt werden, dass sie für die neurologische Entwicklung essenziell sind.[130]
Sulfatierung von Peptiden und Proteinen
[Bearbeiten | Quelltext bearbeiten]
Die Sulfatierung von Tyrosin-Einheiten in Peptiden und Proteinen ist eine weitverbreitete post-translationale Modifikation mit wichtigen biologischen Funktionen.[131] Während die Phosphorylierung vorwiegend Serin- und Threonin-Einheiten betrifft und kaum Tyrosin-Einheiten, betrifft die Sulfatierung hauptsächlich Tyrosin-Einheiten und ist bei diesen vergleichsweise häufig.[132] Katalysiert wird die Tyrosin-Sulfatierung durch eine membrangebundene Sulfotransferase. Tyrosin-Sulfatierung kommt sowohl in Wirbeltieren als auch in Wirbellosen wie Weichtieren und Gliederfüßern vor, nicht aber in Mikroorganismen. Die Sulfat-Gruppen steuern die Wechselwirkungen mit anderen Peptiden und Proteinen.[133] Essenziell ist die Modifikation von Tyrosin bei dem Peptidhormon Cholecystokinin, das die Verdauung reguliert und dessen Sulfatform eine 250-mal höhere Wirkung hat. Das Thyroglobulin, das der Biosynthese der Schilddrüsenhormone in Wirbeltieren dient, weist neben diversen anderen post-translationalen Modifikationen Sulfat-Gruppen auf.[110] Die Bindung von Fibronectin an Fibrin ist ein wichtiger Prozess in der Wundheilung, da sie bei der Beschädigung von Blutgefäßen Gerinnsel bildet. Diese verhindern einerseits den weiteren Blutverlust und stellen andererseits einen Ansatzpunkt für die Reparatur des Gewebes dar.[134] Fibronectin weist Tyrosin-Sulfatierungen auf und ihr Fehlen führt zu einer deutlich schlechteren Bindung an Fibrin.[132]
Weitere tierische Naturstoffe
[Bearbeiten | Quelltext bearbeiten]


Sulfatierte Naturstoffe kommen ansonsten vorwiegend in Meerestieren vor. Seegurken der Familie Holothuriidae verfügen über Giftstoffe, die als Holothurine bezeichnet werden. Diese sind auf der Körperoberfläche und in den Cuvierschen Schläuchen vorhanden, die der Verteidigung dienen. Zusätzlich werden sie ins Wasser abgesondert und sind tödlich giftig für Fische, wodurch die Seegurken gut gegen Fressfeinde geschützt sind.[135] Die Holothurine sind biologisch aktive Triterpenglycoside. Sie dienen einerseits zur Verteidigung gegen Fressfeinde, andererseits als Regulatoren, um die Reifung von Eizellen zu synchronisieren. Sie können Zellmembranen schädigen, worauf ihre hämolytische und fungizide Wirkung beruht.[136] Das Holothurin A verursacht eine irreversible Blockade neuromuskulärer Synapsen. Die negative Ladung durch die Sulfat-Gruppe ist ein essenzielles Element für die biologische Aktivität der Verbindung. Eine Studie an Mäusen zeigte für ein desulfatiertes Analogon bei gleicher Konzentration eine zehnfach schwächere Wirkung, die zudem teilweise reversibel war.[137] Holothurin A kommt in Actinopyga agassizii vor.[138] Das verwandte Holothurin A2 kommt in Holothuria edulis vor.[139] In Seegurken kommen daneben Alkylsulfate wie Octylsulfat[S 4] und Decylsulfat[S 5] vor.[140] Auch in Seescheiden wurden Alkyl- und Alkenylsulfate nachgewiesen, zum Beispiel Isooctylsulfat[S 6] in der roten Seescheide (Halocynthia papillosa).[141] Seesterne bilden Steroidglycoside (Saponine), die als Asterosaponine bezeichnet werden und teilweise zusätzlich mit Sulfat-Gruppen modifiziert sind. Zu den Seesternen, in denen sulfatierte Saponine nachgewiesen wurden, gehören der Nordpazifische Seestern (Asterias amurensis), der Asterosaponin A bildet,[142] und Aphelasterias japonica.[143] Bei der Art Patiria pectinifera wurde nachgewiesen, dass Asterosaponine aus Cholesterol und Cholesterolsulfat[S 7] biosynthetisiert werden.[144] Ein Beispiel für eine Sulfat-Verbindung aus einem landlebenden Tier ist das Zetekitoxin[S 8] aus dem Panama-Stummelfußfrosch (Atelopus zeteki). Es handelt sich um ein Alkaloid, das eine Sulfat-Gruppe aufweist und zusammen mit Tetrodotoxin auftritt.[145] Beide Verbindungen ähneln sich strukturell und ihre Giftwirkung beruht auf der Hemmung spannungsabhängiger Natriumkanäle.[146]
Vorkommen in Pflanzen
[Bearbeiten | Quelltext bearbeiten]Glucosinolate
[Bearbeiten | Quelltext bearbeiten]

Glucosinolate sind pflanzliche Sekundärmetaboliten, die sich von Aminosäuren ableiten und eine Sulfat-Gruppe tragen. Sie kommen in der Ordnung der Brassicales vor, insbesondere in der Familie der Kreuzblütler (Brassicaceae). Glucosinolate sind eine bedeutende Stoffgruppe, die einerseits der Verteidigung (zum Beispiel gegen Insekten) dient, andererseits aber auch für den charakteristischen Geschmack vieler Speisepflanzen aus der Familie verantwortlich ist.[147][148] Zu diesen Speisepflanzen gehören viele Kreuzblütler wie Weiß- und Rotkohl, Brokkoli, Grünkohl, Blumenkohl, Rosenkohl, Pak Choi, Raps sowie Gartenkresse und Brunnenkresse, aber auch Senf und Meerrettich, für deren Schärfe die aus den Glucosinolaten entstehenden Isothiocyanate verantwortlich sind. Zu den Speisepflanzen aus anderen Familien der Ordnung Brassicales, die ebenfalls Glucosinolate enthalten, gehören Kapern und Papaya.[148][149] In den Brassicales machen die Glucosinolate zum Teil einen Anteil am Trockengewicht der Pflanzen um 1 % aus.[148] Stand 2018 waren knapp 90 Glucosinolate bekannt, die vollständig und eindeutig charakterisiert waren.[16] In der Gartenkresse kommen Glucotropaeolin (mit einer Benzyl-Seitenkette) und Gluconasturtiin (mit einer Phenylethyl-Seitenkette) vor.[150]

Die variable Seitenkette wird in der Biosynthese als Erstes aufgebaut und jeweils durch eine Aminosäure bestimmt, die Ausgangspunkt der Biosynthese ist. Zu diesen Ausgangs-Verbindungen gehören Alanin, Leucin, Isoleucin, Valin, Phenylalanin, Tyrosin und besonders häufig Methionin oder Tryptophan.[147][149] Direkter Vorläufer ist aber oft eine kettenverlängerte Aminosäure, beispielsweise Homomethionin[S 9] oder Homophenylalanin.[S 10] Der Strukturteil, der allen Glucosinolaten gemeinsam ist, besteht aus einem Thiohydroxyiminoester, der am S-Atom zusätzlich eine Glucose-Einheit trägt und am O-Atom eine Sulfat-Einheit.[151] Dieser gleichbleibende Teil wird in der Biosynthese nach dem Vorläufer der Seitenkette aufgebaut. Intermediate sind dabei ein Oxim und eine Thiohydroxyiminosäure. Das Schwefelatom wird durch die Aminosäure Cystein zur Verfügung gestellt. Die letzten beiden Schritte der Biosynthese sind die Glycosylierung (Einführung der Glucose-Einheit) und die Sulfatierung (Einführung der Sulfat-Einheit).[149]
In den Brassicaceae liegt ein nennenswerter Teil des vorhandenen Schwefels in Form von Glucosinolaten vor. Der Gehalt an Schwefel ist in dieser Pflanzenfamilie höher als in anderen Pflanzen und sie benötigen für ein gesundes Wachstum eine erhöhte Zufuhr im Vergleich zu anderen Pflanzen. Die Menge an biosynthetisierten Glucosinolaten hängt auch mit der verfügbaren Konzentration an Schwefel im Boden zusammen. Zum Teil kommen im Wurzelbereich von Kreuzblütlern symbiotische Mikroorganismen vor, die elementaren Schwefel oxidieren und als Sulfat verfügbar machen können.[152]
Die Glucosinolate selbst sind biologisch kaum aktiv.[153] Wird die Pflanze jedoch beschädigt, kommen die Glucosinolate in Kontakt mit dem separat vorliegenden Enzym Myrosinase, das den Abbau der Verbindungen katalysiert, wobei die eigentlich wirksamen Stoffe, vorwiegend Nitrile und Isothiocyanate, freigesetzt werden.[147][153] Der Abbau beginnt durch die Deglycosylierung, das heißt die Entfernung der Glucose-Einheit, durch die Myrosinase. Dadurch wird das jeweilige Molekül instabil und zerfällt durch intramolekulare Eliminierung von Sulfat, wobei ein Isothiocyanat entsteht.[151][153] Die Sulfat-Gruppe ist als Strukturelement notwendig, damit die Glucosinolate durch eine Myrosinase abgebaut werden können, Glucosinolat-Derivate ohne Sulfat-Gruppe werden durch das Enzym nicht erkannt.[153] In bestimmten Fällen werden statt Isothiocyanaten entweder Nitrile oder Thiocyanate freigesetzt.[151]


Glucosinolate dienen der Verteidigung. Ihre Abbauprodukte (insbesondere Isothiocyanate) wirken abschreckend, wachstumshemmend oder sogar giftig auf Fressfeinde und Pathogene der Pflanzen, inklusive Säugetiere, Vögel, Insekten, Mollusken, Bakterien und Pilze.[149][151] Für bestimmte Insekten, die auf glucosinolat-haltige Pflanzen als Nahrungsquelle oder für die Eiablage spezialisiert sind, dienen Glucosinolate und ihre Abbauprodukte als Kairomone (chemische Erkennungsmerkmale), um Wirtspflanzen zu finden. Einige Insekten können sogar Glucosinolate sequestrieren, das heißt aufnehmen und einlagern. Diese dienen ihnen dann als Abwehr gegen Vögel und Eidechsen. Beispiele sind die Harlekin-Wanze (Murgantia histrionica) und die Kohlblattlaus (Brevicoryne brassicae). Letztere verfügt über eine eigene Myrosinase zur Aktivierung von Glucosinolaten.[149] Auch gibt es Pilze und Insekten, die durch entsprechende Anpassungen die Verteidigung umgehen können.[151] Die Kohlmotte ist auf Kreuzblütler als Nahrungspflanzen spezialisiert und verfügt über einen Mechanismus zur Inaktivierung der Glucosinolat-Verteidigung dieser Pflanzen durch Entfernung der Sulfat-Gruppen mittels der Glucosinolat-Sulfatase, was eine Freisetzung von Giftstoffen durch die Myrosinase verhindert.[153]
Flavonoidsulfate
[Bearbeiten | Quelltext bearbeiten]

Sulfate von Flavonoiden sind bei bedecktsamigen Pflanzen weitverbreitet und kommen in mindestens 250 Arten aus 32 Familien vor, unter anderem in den Familien der Korbblütler (Asteraceae), der Süßgräser (Gramineae) und der Palmengewächse (Palmae).[154] Als erstes Flavonoidsulfat wurde 1937 das Persicarin (Isorhamnetinsulfat) aus Wasserpfeffer (Polygonum hydropiper) isoliert.[17] Schon 1988 waren über 100 Vertreter der Gruppe bekannt, darunter besonders viele, die sich von dem Flavonol Quercetin oder den Flavonen Apigenin und Luteolin ableiten. Die Verbindungen liegen natürlicherweise als Kalium-, Natrium- oder Calciumsalz vor.[154] Flaveria bidentis (Tageteae) enthält viele Flavonoid-Sulfate, darunter Derivate von Quercetin und Isorhamnetin mit einer bis vier Sulfat-Gruppen.[155] Die Biosynthese von Flavonoidsulfaten in Flaveria chloraefolia wurde detailliert untersucht. Die Art enthält Sulfotransferasen, die Flavonole an spezifischen Positionen sulfatieren können. So werden sie zunächst durch eine Sulfotransferase in Position 3 sulfatiert. Aus Quercetin wird so Quercetin-3-O-sulfat. Flavonol-3-sulfate werden durch andere Enzyme in weiteren spezifischen Positionen sulfatiert zu Flavonol-3,3‘- und Flavonol-3,4‘-sulfaten.[156] Einige sulfatierte Flavonoide sind gleichzeitig auch Glycoside. Die Theograndine sind zwei Glucoside, die in Cupuaçu (Theobrona grandiflorum) vorkommen. Sie leiten sich von Isoscutellarin[S 11] und Hypolaetin[S 12] ab und unterscheiden sich demnach nur in einer Hydroxy-Gruppe. Beide tragen eine Sulfat-Gruppe an der Glucose-Einheit.[157]
Weitere Vorkommen in Pflanzen
[Bearbeiten | Quelltext bearbeiten]
In den Korbblütlern, insbesondere in der Unterfamilie Cichorioideae, kommen sulfatierte Sesquiterpenlactone vor. Dazu gehören die Sulfoscorzonine in Scorzonera divaricata (Gattung Schwarzwurzeln)[158] sowie Derivate des Lactucins[S 13] im Milchsaft des Gartensalats, etwa das 15-Desoxylactucin-8-sulfat.[159] Das Atractylosid ist ein pflanzliches Gift, das für grasende Weidetiere gefährlich ist. Es handelt sich um ein Terpenoid-Glycosid, das an der Glucose-Einheit zusätzlich sulfatiert ist. Die Giftwirkung beruht auf der Störung des Citratzyklus. Zuerst isoliert wurde es 1873 aus dem Gummi-Spindelkraut (Atractylis gummifera). Später wurde die Verbindung auch in anderen Pflanzen auf unterschiedlichen Kontinenten nachgewiesen. Dazu gehören Atractylis carduus (aus der gleichen Gattung) sowie Arten der Gattung Wedelia, die gewöhnliche Spitzklette und Kaffeebohnen.[14] Sulfate von 1-O-Coumaroylglucose[S 14] und 1-O-Caffeoylglucose[S 15] kommen ebenfalls in mehreren Pflanzenarten vor, unter anderem im Adlerfarn. Sulfate des 1-Caffeoylglucosids wurden außerdem im gewöhnlichen Frauenhaarfarn und im Milzfarn nachgewiesen.[160] In Koriander kommen Monoterpen-Derivate mit jeweils drei Alkohol-Gruppen vor. Ein Großteil dieser Verbindungen liegt glykosidisch vor und trägt an einer Alkohol-Gruppe eine Glucose-Einheit. Teilweise ist die Glucose-Einheit zusätzlich mit einer Sulfat-Einheit modifiziert.[161] Die Carrageene sind eine Gruppe von Polysacchariden, die in großer Menge in Rotalgen vorkommen, zum Beispiel im Knorpeltang (Chondrus crispus). Die Struktur ist linear, besteht überwiegend aus Galactose-Einheiten und ist mit einer variablen Anzahl an Sulfat-Gruppen modifiziert.[162]
Vorkommen in Mikroorganismen
[Bearbeiten | Quelltext bearbeiten].jpg)

Mikroorganismen bilden sulfatierte Naturstoffe. Auch sogenannte Muschelgifte, die in Meeresfrüchten vorkommen, werden durch Mikroorganismen synthetisiert. Zu dieser Gruppe gehören die Gonyautoxine, eine Gruppe von giftigen sulfatierten Naturstoffen aus marinen Dinoflagellaten. Ihre Giftwirkung basiert auf einer Hemmung spannungsabhängiger Natriumkanäle.[163] Das Yessotoxin gehört ebenso zu dieser Gruppe und wurde als Verunreinigung in Grünlippmuscheln nachgewiesen. Die Verbindung weist zwei Sulfat-Gruppen auf und wird unter anderem von den Dinoflagellaten Protoceratium reticulatum und Gonyaulax spinifera (Gattung Gonyaulax) produziert.[164][165] Das Maitotoxin ist ein hochgiftiger Naturstoff aus dem Dinoflagellaten Gambierdiscus toxicus. Es hat eine Molmasse von über 3400 g/mol, enthält 164 Kohlenstoffatome sowie zwei Sulfat-Gruppen und gehört zu den größten und giftigsten Naturstoffen, die keine Polymere (zum Beispiel Proteine) sind.[166][167] Cylindrospermopsin[S 16] ist ein sulfatiertes leberschädigendes Toxin, das von Cyanobakterien produziert wird. Es war verantwortlich für eine Reihe von Vergiftungsfällen 1979 in Australien.[168] Die Biosynthese des natürlichen Antibiotikums Ficellomycin aus Streptomyces ficellus verläuft über ein Sulfat. Dabei wird eine Hydroxymethyl-Gruppe an einem Pyrrolidin-Ring durch eine Sulfotransferase mittels Phosphoadenosinphosphosulfat in ein Sulfat umgewandelt. Durch einen intramolekularen nucleophilen Angriff des Stickstoffatoms des Pyrrolidins wird das Sulfat abgespalten und ein Aziridin-Ring gebildet.[169]
Bestimmte Arten von Bakterien und Archaeen können Schwefel-Verbindungen wie Schwefelwasserstoff oder Thiosulfat zur Energiegewinnung verwerten, wobei der enthaltene Schwefel meist zu Sulfat oxidiert wird.[170] Bei phototrophen Bakterien, die Energie aus dem Sonnenlicht ziehen, dient unter anderem Schwefelwasserstoff als Reduktionsmittel zur Gewinnung organischer Verbindungen aus Kohlenstoffdioxid. Der Schwefelwasserstoff wird dabei zunächst zu elementarem Schwefel und dann weiter zu Sulfat oxidiert. Zu diesen Bakterien gehören die Familien Chromatiaceae und Rhodospirillaceae.[171] Bestimmte Bakterien können auch sulfidische Minerale wie Pyrit oxidieren, wodurch sie Sulfat bilden und zur Verwitterung der Minerale beitragen.[172]
Andererseits ist die Nutzung von Sulfat als Elektronenakzeptor, die sogenannte Sulfatatmung, ein Stoffwechselweg bei anaeroben Prokaryoten, sowohl Bakterien als auch Archaeen. Die Sulfatatmung ist vermutlich ein evolutionär alter Mechanismus und ist weitverbreitet, wobei die Gene für die Enzyme der Sulfatatmung eine große Ähnlichkeit aufweisen, selbst bei genetisch weit voneinander entfernten Organismen. Sulfatatmende Mikroorganismen kommen in unterirdischen, sauerstofffreien, Wasser führenden Gesteinsschichten vor.[173] Ein großer Lebensraum solcher Bakterien ist das Schwarze Meer, dessen Sedimente und untere Wasserschichten frei von Sauerstoff sind und große Mengen an Schwefel-Verbindungen enthalten. Viele der dort lebenden Bakterien gehören zur Familie Desulfobaceraceae. Die Umsetzung organischer Verbindungen verläuft praktisch ausschließlich über Sulfatatmung und Methanogenese.[174] Im Großen Salzsee in Utah in den USA wurden in anoxischen (sauerstofffreien) Bereichen ebenfalls Bakterien nachgewiesen, die Sulfat reduzieren, zum Beispiel aus der Gattung Desulfohalobium.[175] Diese Bakterien verwenden Sulfat und Thiosulfat als Elektronenakzeptor zur Energiegewinnung.[176] Sulfat-reduzierende Mikroorganismen setzen bevorzugt leichtere Schwefelisotope um (Isotopeneffekt), vor allem 32S-Sulfat gegenüber 34S-Sulfat, sodass die Isotopenzusammensetzung von Schwefel-Verbindungen zum Teil Aufschluss über die Entstehung geben kann.[177] Sulfat-reduzierende Bakterien kommen außerdem in der menschlichen Darmflora vor. Die Mehrheit der sulfat-reduzierenden Darmbakterien gehören zur Gattung Desulfovibrio.[178]
Abbau und Herstellung
[Bearbeiten | Quelltext bearbeiten]Viele Metallsalze der Schwefelsäure wie Gips und Baryt kommen natürlich vor und werden bergmännisch gewonnen. Daneben können sie auch durch Reaktion mit Schwefelsäure aus elementaren Metallen oder aus Metallsalzen wie Carbonaten hergestellt werden. Für die Herstellung von Schwefelsäure-Estern ist eine größere Zahl an Synthesemethoden bekannt. Zu den wichtigsten Reagenzien in diesem Bereich gehören das Schwefeltrioxid (Anhydrid der Schwefelsäure) und seine Komplexe. Daneben kommen Schwefelsäure und ihre Derivate zum Einsatz.
Gewinnung und Herstellung von Schwefelsäure-Salzen
[Bearbeiten | Quelltext bearbeiten]
Wenige Sulfatminerale werden in großen Mengen in natürlichen Vorkommen abgebaut. Dazu gehören vor allem Calciumsulfat in der Form von Anhydrit und Gips[179] sowie Baryt (Bariumsulfat).[180] Magnesiumsulfat wird meist aus Mischsalzen gewonnen, die als Erze vorkommen, darunter Kieserit, Kainit und Langbeinit.[181]
Metallsulfate können durch Umsetzung der elementaren Metalle mit Schwefelsäure gewonnen werden oder durch Umsetzung geeigneter Salze mit Schwefelsäure, insbesondere Oxide und Carbonate. So wird Chrom(III)-sulfat durch Erhitzen von Chromit oder metallischem Chrom mit Schwefelsäure gewonnen:[182]

Kupfersulfat wird primär durch Umsetzung von Kupfer(II)-oxid mit Schwefelsäure gewonnen:[183]

Eisen(II)-sulfat kann durch Auflösen von Eisen in Schwefelsäure gewonnen werden.[184] Lithiumsulfat wird so aus Lithiumcarbonat gewonnen,[185] Mangansulfat aus Mangancarbonat oder Mangan(II)-oxid,[186] Nickelsulfat aus elementarem Nickel oder Nickel(II)-oxid.[187] Kaliumsulfat kann durch Umsetzung von Kaliumchlorid mit Schwefelsäure gewonnen werden. Für einen rentablen Prozess muss der gleichzeitig anfallende Chlorwasserstoff sinnvoll eingesetzt werden. Ein alternatives Verfahren ist die Umsetzung von Kaliumchlorid mit Magnesiumsulfat, die unter geeigneten Bedingungen – Temperatur und Mischungsverhältnisse – ebenfalls Kaliumsulfat ergibt.[188] Kaliumalaun und Ammoniumalaun werden durch Umsetzung von Aluminiumhydroxid mit Schwefelsäure und Kaliumsulfat oder Ammoniumsulfat hergestellt.[9]
Gips entsteht als Nebenprodukt bei der Herstellung von Titandioxid und in anderen Prozessen.[189] Viele Säuren werden durch Umsetzung ihrer Calciumsalze mit Schwefelsäure hergestellt, wobei Calciumsulfat in Form von Gips als Nebenprodukt entsteht.[179] Ein Großteil der weltweit genutzten Phosphorsäure wird hergestellt, indem Fluorapatit mit Schwefelsäure umgesetzt wird.[190] Analog entsteht Calciumsulfat durch die Herstellung von Fluorwasserstoff aus Fluorit (Calciumfluorid) und die Herstellung von Zitronensäure, Weinsäure und Oxalsäure.[179] Da Kupfererze oft Anteile von Nickel enthalten, fällt Nickelsulfat zum Teil als Nebenprodukt bei der Kupfergewinnung an.[187]
Herstellung von Schwefelsäure-Estern
[Bearbeiten | Quelltext bearbeiten]Für die Herstellung von Schwefelsäure-Estern sind mehrere Verfahren bekannt. Die direkte Veresterung von Alkoholen mit Schwefelsäure ist von geringer Bedeutung. Weitverbreitet sind Umsetzungen mit Schwefeltrioxid, entweder direkt oder in Form von Komplexen mit organischen Verbindungen. Die Handhabung dieser Komplexe ist einfacher und ihre Reaktivität kann je nach Bedarf eingestellt werden. Die Sulfat-Gruppe ist vergleichbar mit der tert-Butyl-Gruppe, was den Raumbedarf anbelangt. Dadurch haben die sterischen Verhältnisse im Molekül einen erheblichen Einfluss darauf, wie leicht sich eine Sulfat-Gruppe einführen lässt.[21]
Umsetzung mit Schwefelsäure
[Bearbeiten | Quelltext bearbeiten]
Die direkte Veresterung von Alkoholen mit Schwefelsäure ist möglich, ergibt aber je nach Edukt oft schlechte Ausbeuten. Einige Edukte, darunter Cellulose, lassen sich dagegen gut mit Schwefelsäure verestern.[191] Schwefelsäure kann durch Dicyclohexylcarbodiimid (DCC) aktiviert werden, wodurch bei der Veresterung von Alkylalkoholen sowie von Phenolen und Oximen gute Ausbeuten erreicht werden können. Nachteilig ist hierbei, dass oft ein großer Überschuss an DCC eingesetzt werden muss, sodass anschließend auch eine große Menge des Nebenprodukts Dicyclohexylharnstoff abgetrennt werden muss. Die Veresterung mit Aktivierung durch DCC wurde zum Beispiel für die Sulfatierung von Kohlenhydraten eingesetzt.[191] Säureempfindliche Edukte können nicht direkt mit Schwefelsäure verestert werden.[192]
Das Reaktionsverhalten bei der Veresterung mit Schwefelsäure/DCC in Dimethylformamid als Lösungsmittel hängt von der Konzentration der Lösung ab. Wird in vergleichsweise verdünnter Lösung gearbeitet, werden nur aliphatische Hydroxy-Gruppen verestert. In konzentrierter Lösung reagieren auch Phenole. Dies ermöglicht zum Teil die selektive Sulfatierung einer von mehreren OH-Gruppen, wie beim Estradiol.[193] Ist die Erzielung einer solchen Selektivität nicht nötig, ist die Reaktion in konzentrierterer Lösung vorteilhaft. Bei 1-Octanol, 1-Tetradecanol und Cyclohexanol werden so bessere Ausbeuten erzielt. Phenol wird in verdünnter Lösung sogar kaum bis gar nicht umgesetzt.[194]
Schwefelsäure kann an die Doppelbindungen von Alkenen addiert werden, wodurch Monoalkyl- und Dialkylsulfate entstehen.[195] So ist die Addition von Schwefelsäure an zwei Moleküle Ethylen eine Methode, um Diethylsulfat herzustellen.[196] Bei der Reaktion von Butenen mit Isobutan zur Herstellung von Alkylatkraftstoff kann als Katalysator Schwefelsäure verwendet werden. In diesem Fall wird in einem Zwischenschritt der Reaktion die Schwefelsäure an die Butene addiert.[197]

Umsetzung mit Schwefelsäure-Derivaten
[Bearbeiten | Quelltext bearbeiten]Tetrabutylammoniumhydrogensulfat, Amidosulfonsäure, Sulfurylchlorid und Chlorsulfonsäure sind Derivate der Schwefelsäure, die sich zur Herstellung von Schwefelsäure-Estern eignen.
Analog zur Veresterung mit Schwefelsäure ist die Veresterung mit Tetrabutylammoniumhydrogensulfat und Dicyclohexylcarbodiimid möglich. Diese Methode wurde zur Herstellung von Flavonoidsulfaten wie aus Apigenin, Luteolin, Quercetin, Rhamnetin und Isorhamnetin eingesetzt.[198][199][200] Die Umsetzung primärer Alkohole mit Amidosulfonsäure ergibt Schwefelsäure-Monoester als Ammoniumsalz.[201] Amidosulfonsäure ist vergleichsweise teuer und wenig reaktiv, wurde aber für die Veresterung primärer gesättigter Alkohole sowie von Kohlenhydraten eingesetzt, wobei oft Katalysatoren wie Harnstoff eingesetzt werden.[191] Amidosulfonsäure eignet sich zur Sulfatierung von Galactomannanen in Dioxan in Gegenwart von Harnstoff.[202] Mit Pyridin als Katalysator wurde Amidosulfonsäure zur Sulfatierung von Flavonoiden eingesetzt, darunter Apigenin, Luteolin, Kaempferol und Quercetin. So kann Quercetin zu Quercetin-3'-O-sulfat umgesetzt werden.[200]

Sulfate können durch Umsetzung von Sulfurylchlorid mit entsprechenden Alkoholen oder Phenolen hergestellt werden. Die Reaktion von Brenzcatechin mit Sulfurylchlorid in Gegenwart von Pyridin ergibt ein cyclisches Sulfat. Ebenso können aus Propanol Dipropylsulfat und aus Butanol Dibutylsulfat hergestellt werden.[203] Die direkte Umsetzung von Diolen mit Sulfurylchlorid zur Synthese cyclischer Sulfate ergibt nur mäßige Ausbeuten, selbst bei sehr niedrigen Temperaturen um −90 °C.[204]
Die Umsetzung von Chlorsulfonsäure mit Alkoholen ergibt Monoester der Schwefelsäure. Werden diese über Natriumsulfat destilliert, entstehen Dialkylsulfate. Dieses Verfahren ist eine Möglichkeit zur Herstellung von Diethylsulfat.[205] Chlorsulfonsäure wurde außerdem zur Sulfatierung von Isoflavonoiden wie Daidzein und Genistein verwendet.[200]
Eine Methode aus dem Jahr 2023 basiert auf einer Kombination aus Tetrabutylammoniumhydrogensulfat und entweder Dimethylsulfat oder Diisopropylsulfat. Diese Dialkylsulfate, die meistens zur Übertragung von Alkyl-Gruppen dienen, können unter geeigneten Bedingungen auch eine Umesterung eingehen. Dabei wird ihre Sulfat-Gruppe auf einen Alkohol, ein Phenol oder ein Kohlenhydrat übertragen. Die beiden Sulfate weisen unterschiedlich große Substituenten auf, wodurch sich in Verbindungen mit mehr als einer Hydroxy-Gruppe je nach eingesetztem Sulfat eine selektive Reaktion erzielen lässt.[206]
-
 Amidosulfonsäure
Amidosulfonsäure -
 Sulfurylchlorid
Sulfurylchlorid -
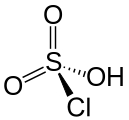 Chlorsulfonsäure
Chlorsulfonsäure



Umsetzung mit Schwefeltrioxid und seinen Komplexen
[Bearbeiten | Quelltext bearbeiten]Dimethylsulfat kann aus Dimethylether und Schwefeltrioxid (SO3) hergestellt werden; die Reaktion ergibt Dimethylsulfat in guter Ausbeute und Reinheit.[21][205] Analoge Reaktionen für Schwefelsäure-Ester mit längeren Alkylketten funktionieren oft schlecht, da solche Alkyl-Gruppen bei der Reaktion oxidiert werden.[205]

Ein verbreitetes Verfahren zur Veresterung von Alkoholen zu Sulfaten ist deren Umsetzung mit Schwefeltrioxid, oft in Form eines Komplexes, beispielsweise mit Dioxan. Einige Alkohole können durch direkte Reaktion mit Schwefeltrioxid verestert werden. Die Reaktion von Methanol mit gasförmigem Schwefeltrioxid oder mit Schwefeltrioxid in Tetrachlormethan ergibt das Monomethylsulfat.[S 17] Destillation von Monomethylsulfat ergibt Dimethylsulfat. Diese Methode wurde historisch für die kommerzielle Gewinnung von Dimethylsulfat eingesetzt. Fettalkohole wie 1-Dodecanol oder 1-Tetradecanol können analog durch Schwefeltrioxid, entweder gasförmig oder gelöst in Schwefeldioxid, sulfatiert werden. Reines Schwefeltrioxid ist allerdings hochreaktiv und schwierig in der Handhabung, da es leicht polymerisiert, was oft zu schlecht reproduzierbaren Resultaten führt. Als Lewis-Säure kann Schwefeltrioxid mit einer Lewis-Base einen Elektronen-Donor-Akzeptor-Komplex bilden. Die Koordinierung an das Schwefeltrioxid erfolgt dabei über ein freies Elektronenpaar der Base. Der Einsatz als Komplex ermöglicht eine einfachere Handhabung und eine Einstellung der Reaktivität.[21] Schwefeltrioxid bildet so vergleichsweise stabile Komplexe, die als Feststoffe vorliegen, u. a. mit Pyridin, Trimethylamin, Triethylamin und Dimethylformamid.[191]


Vergleichsweise starke Basen wie Trimethylamin oder Triethylamin führen zu einer geringeren Reaktivität, schwächere Basen wie Pyridin zu einer höheren Reaktivität, obwohl diese immer noch deutlich geringer ist als bei freiem Schwefeltrioxid. Schwefeltrioxid-Pyridin ist eine stabile Verbindung, die kommerziell erhältlich ist, und eignet sich zur Sulfatierung von Alkoholen, inklusive Sterolen und Kohlenhydraten. Eine Isolierung ist aber nicht immer nötig. Ein Reagenz für die Herstellung von Sulfaten kann ebenso zubereitet werden, indem Chlorsulfonsäure in einen Überschuss an Pyridin gegeben wird. Schwefeltrioxid-Dioxan ist ein deutlich reaktiveres Mittel zur Herstellung von Sulfaten als der Pyridin-Komplex und ist deutlich weniger stabil, sodass es vor der Verwendung frisch zubereitet wird. Schwefeltrioxid-Trimethylamin und Schwefeltrioxid-Triethylamin weisen ähnliche Eigenschaften auf: Sie sind sehr milde Sulfatierungsmittel und können – im Gegensatz zum Pyridin-Komplex – in wässriger Lösung eingesetzt werden. Der Triethylamin-Komplex ist dabei etwas reaktiver. Auch andere Basen wurden schon in SO3-Komplexen eingesetzt, darunter Dimethylanilin und Dimethylformamid.[21] Bei der Reaktion mit einem Schwefeltrioxid-Amin-Komplex wird das Produkt als Ammoniumsalz erhalten, wobei die Stickstoffbase aus dem Komplex das Kation liefert. Eine Frage bei der Auswahl eines Komplexes ist demnach, wie stabil das Produktsalz mit dem entsprechenden Kation ist und wie gut es sich aufreinigen lässt.[191]
Schwefeltrioxid-Dioxan eignet sich zur Sulfatierung vieler Alkohole, wobei die Reaktion oft quantitativ (vollständig) ist. Zu den Alkoholen, die derart umgesetzt werden können, gehören primäre Alkohole wie Ethanol, 1-Butanol und Benzylalkohol, sekundäre wie Cyclohexanol und Borneol, tertiäre wie tert-Butanol und Kohlenhydrate wie Glucose und Galactose. Sterole wie Cholesterol und Ergosterol lassen sich gut mittels Schwefeltrioxid-Pyridin sulfatieren.[21] Die Komplexe mit Trimethylamin und Triethylamin eignen sich für die Sulfatierung aliphatischer Alkohole inklusive Sterolen und Kohlenhydraten.[191] Eine große Zahl an Sterolen wurde erfolgreich mit Schwefeltrioxid-Triethylamin in die entsprechenden Sulfate umgewandelt.[207] Letzteres wurde auch für die Herstellung von Quercetin-Sulfaten verwendet, wobei aber ein Gemisch mehrerer Verbindungen erhalten wurde.[208] Quercetin-3’-O-sulfat wurde gezielt durch Einsatz von Schwefeltrioxid-Dimethylformamid synthetisiert, nachdem bestimmte Hydroxy-Gruppen als Benzylether geschützt waren.[115] Schwefeltrioxid-Dimethylformamid eignet sich daneben zur Sulfatierung von Tyrosin-Einheiten in Peptiden[209] oder zur Herstellung von Sulfaten aus Aminosäuren mit Hydroxy-Gruppen wie Serin und Threonin vor der Synthese von Peptiden daraus.[210] Die vollständige Sulfatierung kleiner Moleküle mit einer größeren Zahl an Hydroxy-Gruppen ist schwierig wegen des geringen Abstands der negativen Sulfat-Gruppen. Solche Reaktionen sind jedoch möglich durch Verwendung von Schwefeltrioxid-Pyridin oder Schwefeltrioxid-Trimethylamin unter Einwirkung von Mikrowellen.[211]

Oxidationsreaktionen
[Bearbeiten | Quelltext bearbeiten]Durch Reaktion eines Alkohols oder Phenols mit Ethylchlorsulfit[S 18] wird ein gemischtes Sulfit gebildet. Dieses kann mit Ruthenium(III)-chlorid und Natriumperiodat zum Sulfat oxidiert werden und die Ethyl-Gruppe mit Natriumiodid abgespalten werden.[191] Die Synthese über Sulfite wird vorwiegend bei cyclischen Sulfaten eingesetzt, bei denen andere Methoden schlechte Ergebnisse liefern. Diese können hergestellt werden, indem zunächst ein cyclisches Sulfit gewonnen und dieses mit Rutheniumchlorid/Natriumperiodat oder mit Ruthenium(VIII)-oxid oxidiert wird. Für die Herstellung cyclischer Sulfite gibt es wiederum mehrere Methoden. Eine ist die Umsetzung eines Epoxids mit Schwefeldioxid und anschließendes Erhitzen. Mit anderen Methoden können größere Ringe erzeugt werden. Mit Diethylaminoschwefeltrifluorid können sowohl 1,2-Diole als auch 1,3-Diole zu cyclischen Sulfiten umgesetzt werden. Bei der Umsetzung eines Diols mit Thionylchlorid oder einem Dialkylsulfit können zusätzlich 1,4-Diole umgesetzt werden.[204] Bei der Elbs-Persulfat-Oxidation wird ein aromatisches Edukt oxidiert und eine vollständige Sulfat-Gruppe eingeführt. Ausgangsprodukt ist ein Phenol, das mit Kaliumperoxodisulfat umgesetzt wird. Die Sulfat-Gruppe wird hierbei gegenüber der Hydroxy-Gruppe des Phenols eingeführt (para-Position) oder, falls diese Position besetzt ist, daneben (ortho-Position). Die Reaktion wird meist dazu genutzt, eine weitere Hydroxy-Gruppe einzuführen, indem das Sulfat hydrolysiert wird.[212] Monomethylsulfat[S 17] kann durch die katalytische Oxidation von Methan mit Oleum hergestellt werden, wobei sich als Katalysator Iod-Verbindungen eignen, inklusive elementarem Iod, Natriumiodid, Kaliumiodid, Iod(V)-oxid oder Kaliumiodat.[213]

Enzymatische Herstellung
[Bearbeiten | Quelltext bearbeiten]
Neben klassischen synthetischen Methoden existieren biotechnologische Methoden zur Herstellung von Schwefelsäure-Estern. Dabei werden Sulfotransferasen eingesetzt, wie sie in Tieren oder Mikroorganismen vorkommen. Sulfotransferasen können zur Sulfatierung zahlreicher Verbindungen eingesetzt werden. Ein Nachteil ist, dass sie als Sulfatquelle überwiegend Phosphoadenosinphosphosulfat (PAPS) benötigen, das einerseits instabil und damit schlecht handhabbar ist, andererseits teuer in der Gewinnung. Zudem werden die Sulfotransferasen durch Phosphoadenosinphosphat (PAP),[S 19] das Abbauprodukt des PAPS, inhibiert, was die Umsetzungen wenig effizient macht. Es sind jedoch Reaktionssysteme bekannt, bei denen zwei Sulfotransferasen zum Einsatz kommen, wobei eine die eigentliche Reaktion katalysiert, während die zweite das PAPS aus PAP regeneriert. Als stöchiometrischer Sulfatüberträger kommt primär para-Nitrophenylsulfat[S 20] zum Einsatz. So muss nur eine geringe Menge an PAP oder PAPS zur Reaktion zugesetzt werden.[214] Bakterielle Sulfotransferasen eignen sich zum Teil dazu, eine SO3-Gruppe direkt von para-Nitrophenylsulfat auf eine Hydroxy-Gruppe zu übertragen. Mit den meisten davon können jedoch nur an Aromaten gebundene (phenolische) Hydroxy-Gruppen sulfatiert werden.[215] Ein typisches Beispiel für diese Enzymgruppe, die einen Phenylester als Sulfatquelle verwenden kann und nur aromatische Hydroxy-Gruppen umsetzt, ist eine Sulfotransferase aus Clostridium innocuum.[216] Ein System aus zwei Sulfotransferasen wurde zur Sulfatierung von Phenol, Brenzcatechin und Biphenyl-4,4'-diol genutzt.[214] Eine Sulfotransferase aus Desulfitobacterium hafniense ist im Gegensatz zu den meisten verwandten Enzymen nicht nur in der Lage, Phenole zu sulfatieren, sondern auch aliphatische Hydroxy-Gruppen, darunter die von 1-Butanol, 1-Pentanol oder 2-Phenylethanol.[215] Ein neueres System verwendet statt para-Nitrophenylsulfat das N-Hydroxysuccinimidsulfat.[S 21] Dessen Reaktionsprodukt, N-Hydroxysuccinimid, ist weniger giftig und leichter vom Produkt abzutrennen als para-Nitrophenol.[217]
Reaktionen
[Bearbeiten | Quelltext bearbeiten]Schwefelsäure-Salze sind wenig reaktiv, lassen sich bei starker Erhitzung jedoch zersetzen und können durch entsprechend drastische Bedingungen reduziert werden. Viele Schwefelsäure-Ester sind hingegen reaktive Verbindungen, die als starke Alkylierungsmittel wirken und sich oft leicht hydrolysieren lassen. Schwefelsäure-Monoester sind als Salze jedoch vergleichsweise stabil.
Reaktionen von Schwefelsäure-Salzen
[Bearbeiten | Quelltext bearbeiten]
Die meisten Metallsulfate zersetzen sich beim Erhitzen, allerdings erst bei einer Temperatur von mehreren hundert Grad. Bei der Reaktion wird Schwefeltrioxid frei und es bleiben Metalloxide zurück. Enthält eine Verbindung Kristallwasser, wird bei der Erhitzung erst dieses abgegeben.[218] Werden Hydrogensulfate wie Natriumhydrogensulfat oder Kaliumhydrogensulfat stark erhitzt, können Hydrogensulfat-Ionen unter Wasserabspaltung zu Pyrosulfat dimerisieren.[219][220] Ab etwa 500 °C zersetzt sich Pyrosulfat unter Abspaltung von Schwefeltrioxid, wobei Sulfat zurückbleibt.[220]
Metallsulfate können außerdem reduziert werden, unter anderem mit Wasserstoff, wobei je nach Kation verschiedene Produkte entstehen. Die Sulfate der Erdalkalimetalle (Calciumsulfat, Strontiumsulfat und Bariumsulfat) sowie Nickelsulfat, Cobaltsulfat und Cadmiumsulfat werden zu den jeweiligen Sulfiden reduziert. Im Falle von Aluminiumsulfat, Magnesiumsulfat und Berylliumsulfat wird ebenfalls das Schwefelatom reduziert, allerdings zum Schwefeldioxid, sodass die Salze in Oxide umgewandelt werden. Im Falle von Eisen(II)-sulfat, Mangansulfat, Zinksulfat und Bleisulfat tritt ein Gemisch von Oxiden und Sulfiden auf. Bei anderen Verbindungen wird stattdessen das Kation reduziert, zum Beispiel von Eisen(III)-sulfat zu Eisen(II)-sulfat und von Kupfer(II)-sulfat über Kupfer(I)-sulfat zu elementarem Kupfer.[221] Natriumsulfat kann auch mit Kohlenstoff reduziert werden.[222] Andererseits können Sulfat-Ionen, beispielsweise in einer Ammoniumsulfat-Lösung, elektrochemisch zu Peroxodisulfat oxidiert werden.[223]
Alkylierungen oder Substitutionsreaktionen
[Bearbeiten | Quelltext bearbeiten]Dialkylsulfate sind starke Alkylierungsmittel. Die Reaktivität ist größer, je kürzer der Alkylsubstituent ist.[224] Die Bildung eines Sulfats kann so zur Aktivierung von Alkoholen dienen, um diese in andere Verbindungen wie Alkylchloride zu überführen.[225] Dialkylsulfate können diverse Verbindungen alkylieren, darunter Phenole, Carbonsäuren und deren Salze, Aromaten und Thiole.

Alternativ zu Diazomethan eignet sich Dimethylsulfat als Reagenz für eine einfache Umwandlung von Carbonsäuren in ihre Methylester. Das Proton der Carbonsäure muss hierbei durch eine geeignete Base wie Dicyclohexylethylamin[S 22] neutralisiert werden. Eine Alternative ist die Deprotonierung der Carbonsäure mit Lithiumhydroxid vor der Umsetzung mit Dimethylsulfat.[226][227] Dimethylsulfat kann daneben für eine Williamson-Ethersynthese eingesetzt werden. Ein Beispiel hierfür ist die Methylierung von Phenol zu Anisol.[228] Dialkylsulfate eignen sich für die Alkylierung von Thiol-Gruppen zu Sulfiden. So kann para-Thiokresol mittels Dimethylsulfat oder Diethylsulfat in das entsprechende Methyl- oder Ethylsulfid überführt werden.[229] Dialkylsulfate können zudem zur Alkylierung von Aromaten wie Benzol verwendet werden. Die Reaktionsbedingungen entsprechen im Wesentlichen einer Friedel-Crafts-Alkylierung, bei der normalerweise eine Lewis-Säure wie Aluminiumchlorid sowie ein Alkylhalogenid als Alkylierungsmittel verwendet wird. Letzteres kann jedoch durch ein Dialkylsulfat ersetzt werden, beispielsweise Dimethylsulfat, Diethylsulfat, Diisopropylsulfat oder Dibutylsulfat.[230] Schließlich können Dialkylsulfate auch Carbonsäureamide zu Iminiumsalzen alkylieren. Ein Beispiel hierfür ist die Methylierung von Dimethylformamid mit Dimethylsulfat.[231]
Über ein cyclisches Sulfat können aus einem Diol andere difunktionalisierte Moleküle hergestellt werden. Der Ring kann durch viele Nucleophile geöffnet werden. Dazu gehören Azid-Ionen, Fluorid-Ionen (aus Tetrabutylammoniumfluorid), Thiocyanat-Ionen, Nitrat-Ionen, Carboxylat-Ionen, Alkoholaten, Thiolaten, Acetyliden, Aminen, Cyanid, Organocupraten oder Hydrid (aus Lithiumaluminiumhydrid).[204][232] Dadurch entsteht ein offener Schwefelsäure-Monoester. Dieser kann hydrolysiert werden, um einen α-funktionalisierten Alkohol zu erhalten, oder mit einem zweiten Nucleophil umgesetzt werden. Ein Beispiel für letzteres ist die Bildung eines Aziridins durch eine intramolekulare Reaktion. Dabei wird das cyclische Sulfat mittels Lithiumazid geöffnet und die Azid-Gruppe durch Lithiumaluminiumhydrid reduziert. Die intramolekulare Substitution ergibt den Ringschluss zum Aziridin.[232] Analog kann durch Umsetzung mit Di-tert-butylmalonat[S 23] und Natriumhydrid ein Cyclopropan-Ring gebildet werden.[204]

Hydrolyse
[Bearbeiten | Quelltext bearbeiten]Schwefelsäure-Diester und -monoester können in wässriger Lösung leicht hydrolysiert werden: Dimethylsulfat kann über Monomethylsulfat[S 17] zu Methanol und Sulfat hydrolysiert werden. Analog wird Ethylensulfat in zwei Schritten zu Ethylenglycol und Sulfat hydrolysiert. Dabei treten unterschiedliche Reaktionsmechanismen auf: Bei der Reaktion eines Diesters zum Monoester wird eine C-O-Bindung gebrochen, das heißt der Schwefelsäure-Monoester ist die Abgangsgruppe. Das Gleiche gilt bei der Hydrolyse eines Monoesters in basischer Lösung. Bei der Hydrolyse eines Monoesters im Sauren hingegen wird eine S-O-Bindung gebrochen, die Abgangsgruppe ist der Alkohol.[233] Monoalkylsulfate sind als Salze, zum Beispiel als Natriumsalze, vergleichsweise stabil. In protonierter Form können sie jedoch selbst in unpolaren, inerten Lösungsmitteln wie Diethylether oder Tetrahydrofuran hydrolysiert werden, wenn diese Spuren von Wasser oder anderen Verunreinigungen enthalten.[234] Monoarylsulfate sind demgegenüber vergleichsweise stabil. Die Hydrolyse von Diphenylsulfat[S 24] zu Monophenylsulfat ohne Weiterreaktion ist problemlos möglich.[235]

Komplexe und Einschluss-Verbindungen
[Bearbeiten | Quelltext bearbeiten]

Metallkomplexe, die Sulfat-Ionen als Liganden tragen, werden als Sulfato-Komplexe bezeichnet. Das Sulfat-Ion kann dabei unterschiedliche Bindungsmodi annehmen: über ein Sauerstoffatom gebunden (monodentat), über zwei Sauerstoffatome an dasselbe Metallatom gebunden (bidentat) oder gleichzeitig an zwei oder mehr Metallatome gebunden (verbrückend). Durch seine vier Sauerstoffatome kann das Ion an bis zu zehn Metallatome gleichzeitig koordinieren. Der Koordinationsmodus in Sulfato-Komplexen kann durch Infrarot-Spektroskopie (IR-Spektroskopie) unterschieden werden. In einem freien Sulfat-Ion sind alle vier S-O-Bindungen identisch. Ist das Ion über ein Sauerstoffatom an ein Metallatom komplexiert (als monodentater Ligand), ist dessen S-O-Bindung nicht mehr identisch zu den anderen drei, was im entsprechenden IR-Spektrum anhand der Streckschwingungen der Bindungen detektiert werden kann. Sulfat kann daneben als bidentater Ligand wirken, das heißt über zwei Sauerstoffatome an ein Metallatom gebunden. In diesem Fall liegen zwei Paare von Bindungen vor, die sich jeweils gleich verhalten, was wiederum mittels IR-Spektroskopie untersucht werden kann.
Sulfat bildet mit Kupfer, Cobalt und anderen Übergangsmetallen stabile, kristallisierbare Komplexe.[236] Ein Beispiel für einen Komplex mit einem monodentaten Sulfat-Liganden ist [Pt(NH3)4(SO4)(NO)]+. In diesem Komplex trägt das zentrale Platin-Atom neben einem Sulfat-Ion auch noch vier Moleküle Ammoniak und ein Nitrosyl-Ion als Liganden.[237] Oft können Sulfato-Komplexe durch Oxidation von Schwefeldioxid hergestellt werden. Der Disauerstoff-Ligand in Sauerstoff-Komplexen ist reaktiver als molekularer Sauerstoff. So können Sauerstoff-Komplexe von Eisen, Nickel, Ruthenium, Rhodium, Palladium, Platin und Iridium jeweils leicht Schwefeldioxid oxidieren, wobei sich ein Sulfato-Komplex bildet. Ein Iridium-Komplex, der eine solche Reaktion eingeht, ist [Ir(PPh3)2(CO)I(O2)], der neben dem Sauerstoff-Liganden je einen Iodid- und Carbonyl-Liganden sowie zwei Triphenylphosphin-Liganden aufweist.[238]
Analog kann als Edukt der Platin-Komplex [Pt(PPh3)2(O2)], verwendet werden. Liegt dieser in Lösung vor, bildet sich beim Einleiten von Schwefeldioxid sofort der Sulfat-Komplex [Pt(PPh3)2(SO4)]2−. Umgekehrt kann diese Verbindung über den Schwefeldioxid-Komplex hergestellt werden. Wird Schwefeldioxid in eine Lösung des Platin-Ethylen-Komplexes [Pt(PPh3)2(C2H4)] geleitet, bildet sich durch Verdrängung des Ethylens der grün gefärbte Komplex [Pt(PPh3)2(SO2)]. Durch Erhitzen des Komplexes an Luft oder Durchleiten von Sauerstoff durch seine Lösung findet eine Oxidation statt und es bildet sich wiederum [Pt(PPh3)2(SO4)]2−.[239] Mit einigen Metallen wie Neptunium bildet Sulfat auch in Lösung vergleichsweise stabile Komplexe.[240] Uranyl-Ionen bilden in Lösung Komplexe mit ein bis drei Sulfat-Ionen.[241]

Organische Moleküle mit bestimmten Struktureinheiten können wiederum an Sulfat-Ionen koordinieren und Einschluss-Verbindungen bilden. Dazu gehören cyclische Verbindungen, in denen Sulfat-Ionen durch elektrostatische Wechselwirkungen festgehalten werden, aber auch solche, die Sulfat-Ionen in allen drei Raumrichtungen umschließen können.[242] Ein solcher Ligand leitet sich vom Harnstoff ab und trägt zusätzlich zwei 2,2’-Bipyridin-Einheiten. Dieser kann durch Ausbildung von Wasserstoffbrücken eine Käfig-Verbindung mit einem Sulfat-Ion bilden, bei der das Sulfat dicht eingeschlossen und von der Umgebung abgeschirmt ist. Die Käfigstruktur besteht dabei aus sechs dieser Liganden und hat erheblichen Einfluss auf die Eigenschaften des eingeschlossenen Ions. So verhindert der Käfig, dass bei der Zugabe von Strontiumnitrat das Sulfat-Ion als Strontiumsulfat ausfällt.[243] Ein anderer bekannter Ligand besteht aus drei zusammenhängenden identischen Einheiten mit insgesamt sechs Harnstoff-Gruppen. Mit diesem Ligand können Sulfat-Ionen sehr effizient aus einer wässrigen Lösung entfernt und in ein organisches Lösungsmittel überführt werden.[244]
Verwendung
[Bearbeiten | Quelltext bearbeiten]Natürlich vorkommende Sulfate dienen oft als Primärrohstoffe, andererseits sind Sulfate Intermediate bei der Aufbereitung anderer Erze. Calciumsulfat ist ein zentrales Produkt der Baustoffindustrie. Daneben haben Salze und Ester der Schwefelsäure verschiedene Anwendungszwecke in der Medizin sowie als Tenside, Farbstoffe, Gerbstoffe und Düngemittel.
Rohstoffgewinnung und chemische Industrie
[Bearbeiten | Quelltext bearbeiten]Verschiedene Sulfatminerale sind industriell relevante Erze und Rohstoffe, insbesondere Gips. Baryt (Bariumsulfat) ist das wichtigste Bariumerz. Es dient als Ausgangsstoff für die Herstellung anderer Barium-Verbindungen, wobei es in der Regel zunächst zu Bariumsulfid reduziert wird.[180] Coelestin (Strontiumsulfat) ist neben Strontianit (Strontiumcarbonat) eines von zwei industriell relevanten Strontiumerzen. Coelestin wird zunächst zu Strontiumcarbonat umgesetzt, aus dem die meisten anderen Strontium-Verbindungen hergestellt werden.[245] Das wichtigste Bleierz ist Galenit (Bleisulfid), die Lagerstätten enthalten aber oft durch Oxidation entstandenes Anglesit (Bleisulfat), das beim Abbau mit verwertet wird.[246]
Aluminiumsulfat ist nach Aluminiumoxid die zweitwichtigste Aluminium-Verbindung in der Industrie und wird als Ausgangsstoff zur Herstellung der meisten anderen Aluminium-Verbindungen verwendet.[9] Ebenso werden Magnesiumsulfat,[186] Nickelsulfat[187] und Kaliumsulfat[188] als Zwischenprodukte gewonnen, um aus Erzen andere Verbindungen herzustellen. Mangansulfat dient zur Herstellung von elementarem Mangan sowie von Manganlinolat[S 25] und verwandten Verbindungen, die als Sikkative eingesetzt werden.[186] Titanylsulfat ist ein Intermediat bei der Herstellung von Titandioxid nach dem Sulfat-Prozess. Hierbei wird das Titanerz Ilmenit mit Schwefelsäure zu Titanylsulfat umgesetzt, dieses hydrolysiert und dann bei 950 °C kalziniert.[247] Bei der Gewinnung von Seltenerdmetallen aus Erzen wird Schwefelsäure zum Aufschluss eingesetzt, wodurch Sulfate als Intermediate entstehen.[248] Die mengenmäßig bedeutendste Verwendung von Bariumsulfat ist in Bohrflüssigkeit in der Erdölgewinnung.[249] Das Bariumsulfat erhöht die Dichte der Flüssigkeit, die dazu dient, Gesteinsmaterial aus dem Bohrloch auszuschwemmen und die Bohrvorrichtung zu kühlen.[250]

Dialkylsulfate eignen sich als Alkylierungsmittel, zum Beispiel für die Umsetzung von Carbonsäuren zu deren Alkylestern oder von Aminen zu quartären Ammonium-Verbindungen. Industriell ist Dimethylsulfat die mit Abstand meistgenutzte Verbindung aus der Gruppe der Dialkylsulfate, lediglich Diethylsulfat hat ebenfalls eine gewisse (wenn auch geringe) Bedeutung. Dimethylsulfat wird unter anderem zur Herstellung von Methylsalicylat aus Salicylsäure verwendet sowie zur Umsetzung von Kaliumiodid zu Methyliodid. Diethylsulfat eignet sich zur Herstellung der Ethylester von Fettsäuren, die als Weichmacher verwendet werden.[224]
Bauwesen
[Bearbeiten | Quelltext bearbeiten]Sulfate spielen eine enorme Rolle im Bauwesen. Gips wird zum Verputzen von Wänden sowie in Form fertiger Platten verwendet.[76][251] Essenziell für die Verwendung als Wandverputz ist, dass Calciumsulfat einerseits leicht durch Erhitzung dehydriert werden kann, dass die Hydrate durch Anmischen mit Wasser aber andererseits leicht wieder zurückgebildet werden.[252] Gipskartonplatten bestehen hauptsächlich aus gehärtetem Gips, der zwischen zwei Schichten Papier zusammengehalten wird.[253] Weitere Verwendungen sind Fertigbauelemente und Deckenplatten. Gips als Baumaterial hat mehrere vorteilhafte Eigenschaften wie vergleichsweise geringes Gewicht, brandhemmende Eigenschaften (durch das enthaltene Kristallwasser) und schallisolierende Eigenschaften.[76]
Calciumsulfate sind außerdem in Zement enthalten, wobei Gips (das Dihydrat) je nach Grad der Erhitzung bei der Produktion zu Calciumsulfat-Hemihydrat oder Anhydrit (ohne Kristallwasser) entwässert wird. Das Calciumsulfat verbessert die Abbindeeigenschaften des Zements, der sonst zu schnell fest wird, was eine praktische Verarbeitung verhindert.[2][254] Die langsamere Aushärtung beruht auf der Bildung von Ettringit in der angerührten Zementmischung.[254] Der Einsatz als Reaktionsverzögerer in Zement macht einen erheblichen Anteil des weltweit gewonnenen Gipses aus.[252] Baryt (Bariumsulfat) ist Bestandteil von Schwerbeton, der zur Abschirmung von Strahlung eingesetzt wird, zum Beispiel in Atomkraftwerken und medizinischen Einrichtungen, aber auch in Betongewichten.[255]
Medizin
[Bearbeiten | Quelltext bearbeiten]
Neben Sulfaten, die als Pharmazeutika verwendet werden, wird Gips (Calciumsulfat) für Gipsverbände zur Behandlung von Knochenbrüchen verwendet. Er ist billig, einfach in der Anwendung und für Röntgenstrahlung durchlässig, was eine Untersuchung von Knochen unter dem Gips ermöglicht. Aufgrund dieser Eigenschaften sind Gipsverbände heute nach wie vor verbreitet in Gebrauch.[12]
Sulfatsalze werden medizinisch vielfältig genutzt. Das meistgenutzte Präparat zur Supplementierung von Eisen bei Eisenmangelanämie ist Eisen(II)-sulfat. Eisenmangelanämie ist eine verbreitete Mangelerkrankung, vorwiegend bei Kindern, und tritt auf, wenn über die Ernährung zu wenig Eisen aufgenommen wird.[256] Bariumsulfat ist ein Kontrastmittel für die Röntgenuntersuchung und die Computertomografie des Gastrointestinaltrakts.[257] Magnesiumsulfat wird zur Behandlung eklamptischer Krämpfe sowie gegen akuten Magnesiummangel eingesetzt.[13] Lithiumsulfat wird gelegentlich in der Lithiumtherapie bei bipolaren Störungen genutzt. Gegenüber dem hauptsächlich genutzten Lithiumcarbonat hat es jedoch nur untergeordnete Bedeutung.[258][259]
Neben den Metallsalzen werden auch organische Wirkstoffe oft als Sulfatsalze eingesetzt. Zur Verbesserung der Eigenschaften, speziell der Wasserlöslichkeit, werden Medikamente oft in eine ionische Form überführt und als Salz eingesetzt. Dabei muss ein geeignetes Gegenion verwendet werden, da dieses ebenfalls Einfluss auf die Eigenschaften der Formulierung hat. Eine im Jahr 2007 publizierte Analyse aller von der FDA in den USA zugelassenen Medikamente ergab, dass ungefähr 38 % ionische Verbindungen waren, bei denen das Kation die aktive Komponente ist. Das mit Abstand häufigste Gegenion war dabei das Chlorid, gefolgt von Sulfat.[260] Das Parasympatholytikum Atropin beispielsweise wird überwiegend als Sulfat (Atropini sulfas, Atropinum sulfuricum, schwefelsaures Atropin) eingesetzt.[261]


Einige Pharmazeutika sind Schwefelsäureester. So kommen Sulfat-Gruppen in einigen Wirkstoffen aus der Gruppe der β-Lactamase-Inhibitoren vor, die zusammen mit β-Lactam-Antibiotika eingesetzt werden. β-Lactam-Antibiotika stören die Peptidoglycansynthese in Bakterien, was zu instabilen Zellwänden und schließlich zum Absterben der Zellen führt.[262] Antibiotika-Resistenzen gegen diese Wirkstoffe sind allerdings weitverbreitet. Ein Mechanismus, der eine solche Resistenz verursacht, ist die Bildung von β-Lactamasen durch die Bakterien. Dabei handelt es sich um Enzyme, die die Amid-Bindung in den β-Lactam-Molekülen spalten und die Antibiotika somit wirkungslos machen können. Dies kann durch den zusätzlichen Einsatz eines β-Lactamase-Inhibitors verhindert werden.[263] Der erste solche Wirkstoff, der zum Einsatz kam, ist das Avibactam.[264] Das Kombinationspräparat mit dem Antibiotikum Ceftazidim wurde im Februar 2015 in den USA zugelassen. Die Kombination kommt gegen verschiedene antibiotikaresistente Pathogene zum Einsatz, darunter entsprechende Stämme von Pseudomonas aeruginosa.[265] Avibactam ist ein kovalenter Inhibitor für verschiedene β-Lactamasen, der mittels seines C7-Carbonyls als Carbamat an ein Serin im aktiven Zentrum bindet. Die Verbindung hat strukturelle Ähnlichkeit zum Ceftazidim, mit dem zusammen sie eingesetzt wird. Die Sulfat-Gruppe des Avibactams mit seiner negativen Ladung entspricht dabei der Carboxylat-Gruppe des Ceftazidims. Durch die Hemmung der β-Lactamase wird ein Abbau des eigentlich wirksamen Antibiotikums verhindert. Die Kombination Ceftazidim-Avibactam wirkt so gegen Bakterien, die β-Lactamasen produzieren und durch Ceftazidim allein nicht abgetötet werden.[262] Durlobactam[S 26] ist eine mit Avibactam strukturell eng verwandte Verbindung, die ähnlich verwendet wird. Ein Kombinationspräparat mit Sulbactam wurde im Mai 2023 in den USA zugelassen und wird für die Behandlung bestimmter β-lactamresistenter Bakterien verwendet, insbesondere resistenter Stämme von Acinetobacter baumannii, das in Bezug auf Antibiotika-Resistenzen zu den problematischsten Pathogenen gehört.[264]

Eine weitere Gruppe medizinisch genutzer Schwefelsäureester sind die konjugierten equinen Estrogene. Dabei handelt es sich um ein Gemisch von Schwefelsäureestern verschiedener Estrogene wie Estron, Estradiol und Equilin. Das Wort equin (von Pferd) bezieht sich dabei auf die Gewinn aus dem Urin trächtiger Stuten. Eingesetzt werden die konjugierten Estrogene bei Wechseljahresbeschwerden, die durch ein Mangel an körpereigenen Estrogenen verursacht werden.[266] Einige Sucralfat ist ein Komplex aus Aluminiumhydroxid und sulfatierter Saccharose und wird in verschiedenen Ländern bei Krankheitsbildern eingesetzt, die mit Magengeschwüren einhergehen. Sucralfat wird kaum resorbiert und bildet eine Schutzschicht, wodurch eine Heilung befördert wird.[267] Das natürlich vorkommende Heparin ist ein sulfatiertes Polysaccharid. Es wird als gerinnungshemmendes Medikament (Antikoagulans) eingesetzt gegen Thrombosen und Lungenembolien. Es wird vorwiegend aus Schweinedärmen extrahiert. Ein synthetisches Oligosaccharid mit analoger gerinnungshemmender Wirkung ist das Fondaparinux. Seine Struktur entspricht genau der Abfolge aus fünf Zuckern, die für die gerinnungshemmende Wirkung des Heparins verantwortlich ist. Fondaparinux ist seit 2003 auf dem Markt.[125]
Farbstoffe und Pigmente
[Bearbeiten | Quelltext bearbeiten]
Reaktivfarbstoffe sind solche Farbstoffe, die beim Färbeprozess mit Textilfasern (oder einem anderen Substrat) kovalente Bindungen ausbilden, und gehören zu den industriell meistgenutzten Farbstoffen. Eine Variante sind Verbindungen, die eine Sulfat-Gruppe als Bestandteil einer β-Sulfatoethylsulfon-Gruppe enthalten wie das Reactive Black 5.[268] Die eigentlich reaktive Gruppe, die die kovalente Bindung ausbildet, ist eine Vinylsulfon-Gruppe. Diese ist als β-Sulfatoethylsulfon-Gruppe maskiert.[269] Beim Erhitzen des Farbstoffs bei hohem pH-Wert während des Färbeprozesses wird Sulfat eliminiert und so die reaktive Gruppe freigesetzt.[269][270] Die Farbstoffe werden durch Veresterung von Hydroxyethylsulfonen mit Schwefelsäure gewonnen und neutralisiert (zum Beispiel mit Natriumhydroxid), um ein Salz zu erhalten. Sie eignen sich für die Färbung von Wolle und Baumwolle.[270] Gefälltes Bariumsulfat dient als Beschichtungspigment in der Papierherstellung und als Füllstoff in Farben, Lacken und Tinte.[249] Lithopone ist ein Weißpigment, das aus Bariumsulfat und Zinksulfid besteht. Hergestellt wird es, indem in Lösung Bariumsulfid mit Zinksulfat gemischt wird.[271] Es wird als Pigment für Farben und in Kunststoff verwendet.[272]
Sonstige Verwendungen
[Bearbeiten | Quelltext bearbeiten]Chrom(III)-sulfat ist der bedeutendste Gerbstoff in der Gerberei. Dabei werden Tierhäute zu Leder verarbeitet, wobei sie derart modifiziert werden, dass sie haltbar werden und eine höhere thermische Stabilität aufweisen, gleichzeitig aber die Flexibilität einer frischen Haut behalten. Chrom(III)-Ionen vernetzen Kollagen-Stränge sowohl durch Wasserstoffbrücken als auch durch kovalante Brücken zwischen den Carboxylgruppen von Asparaginsäure- und Glutaminsäure-Resten. Auf dieser Vernetzung beruht die Haltbarkeit des Materials. Die Koordination von Sulfat-Ionen an die Chrom-Ionen beeinflusst die Reaktivität und führt zu besseren Materialeigenschaften (höhere thermische Stabilität). Andere Gerberei-Reagenzien sind gegenüber Chromsulfat von untergeordneter Bedeutung, zu diesen gehören organische Gerbstoffe und das Aluminiumsulfat.[273]

Die Monoester der Schwefelsäure mit längerkettigen Alkoholen (Fettalkoholen) werden als Fettalkoholsulfate bezeichnet und in Reinigungsmitteln und Hygieneprodukten verwendet. Da sie in freier Form nicht stabil sind, werden sie als Salze, beispielsweise als Natriumsalze, verwendet. Die meistgenutzten Vertreter sind das Natriumlaurylsulfat (vom Laurylalkohol mit zwölf Kohlenstoffatomen) sowie Verbindungen mit 14, 16 oder 18 Kohlenstoffatomen in der Alkylkette. Verwendet werden sie als Tenside in Wasch- und Spülmitteln. Die Alkylethersulfate verfügen zwischen der Sulfat-Gruppe und dem Alkyl-Rest zusätzlich über ein bis vier Ethylenglycol-Gruppen (-CH2CH2O-). Alkylethersulfate wie das Natriumlaurylethersulfat bilden besonders viel Schaum, weshalb sie in Schaumbädern und Shampoos verwendet werden.[274] Neben den Natriumsalzen werden Alkylethersulfate zum Teil als Ammonium- und Magnesiumsalze in Hygieneprodukten verwendet. Dazu gehören Ammoniumlaurylethersulfat[S 27] und Magnesiumlaurylethersulfat.[275][S 28]
Carrageen ist ein natürliches sulfatiertes Polysaccharid, das viel in der Lebensmitteltechnik verwendet wird, unter anderem als Verdickungs- und Geliermittel. Für die Gewinnung von Carrageen werden Rotalgen, insbesondere solche der Gattung Kappaphycus, kommerziell angebaut.[276] Viele Sulfatsalze werden als Dünger eingesetzt, darunter Ammoniumsulfat und das Doppelsalz Ammoniumsulfatnitrat,[277] ebenso das Magnesium-Mineral Kieserit.[278] Auf mangan-armen Böden wird Mangansulfat als Dünger eingesetzt.[186] Der am häufigsten genutzte Kaliumdünger ist Kaliumchlorid. Das teurere Kaliumsulfat wird nur in besonderen Fällen eingesetzt, wenn die Gefahr der Versalzung des Bodens besteht oder bei Pflanzen, die empfindlich auf Chlorid reagieren, wie Tabak und Zitrusfrüchte.[188] Kupfersulfat-Pentahydrat wird in der Landwirtschaft als Fungizid eingesetzt.[279]

Aluminiumsulfat wird hauptsächlich in der Papierherstellung und Wasserreinigung verwendet. In der Papierindustrie dient es zur Fixierung von Schlichtemitteln und Farben sowie als Beschichtung für Glanzpapier. In der Wasseraufbereitung wird es als Flockungsmittel verwendet.[9] Die Hauptverwendung von Nickelsulfat ist die galvanische Vernickelung, das heißt die Beschichtung mit metallischem Nickel.[187] Analog wird Rhodium(III)-sulfat zur Rhodinierung (Beschichtung mit Rhodium) verwendet.[280]
Natriumsulfat und Magnesiumsulfat werden zum Trocknen organischer Lösungsmittel verwendet.[281] Wasserfreies Kupfersulfat eignet sich ebenfalls zum Trocknen organischer Lösungsmittel sowie durch die Farbänderung bei der Hydratbildung als Indikator für die Anwesenheit von Wasser in Lösungsmitteln.[279]
Nachweis
[Bearbeiten | Quelltext bearbeiten]Sulfat kann durch Turbidimetrie quantitativ bestimmt werden, das heißt durch Messung einer Trübung. Dabei wird Bariumchlorid in die sulfathaltige Lösung gegeben, was zur Entstehung von Bariumsulfat führt, welches die Trübung verursacht.[282] Auch in der klassischen Analytik dient die Bildung von Bariumsulfat als Nachweise. Dazu wird die Probelösung in Salzsäure mit Bariumchlorid versetzt:
- Sulfat-Ionen bilden mit Barium-Ionen einen weißen, säure-unlöslichen Niederschlag von Bariumsulfat.

Die Säure wird zur Entstörung zugesetzt, da Carbonat, Phosphat oder Sulfit mit Barium in Wasser ebenfalls schwer lösliche Salze bilden, die im Gegensatz zu Bariumsulfat aber in Salzsäure löslich sind. Wird der Nachweis in Gegenwart von Permanganat durchgeführt, bilden sich violett gefärbte Mischkristalle. Das darin eingebaute Permanganat kann dann nicht mehr einfach mit Reduktionsmitteln reagieren und die Kristalle so nicht entfärbt werden. Ein weiterer Nachweis, der ebenfalls auf der Bildung von Bariumsulfat beruht, verwendet Bariumchlorid und Natriumrhodizonat.[S 29] Aus den beiden Reagenzien wird rotes Bariumrhodizonat[S 30] gebildet, das bei Zugabe von Sulfat durch Bildung von Bariumsulfat wieder entfärbt wird.[283]
Anhang
[Bearbeiten | Quelltext bearbeiten]Literatur
[Bearbeiten | Quelltext bearbeiten]- Charles P. Hoiberg, Ralph O. Mumma: Preparation of sulfate esters. Reactions of various alcohols, phenols, amines, mercaptans, and oximes with sulfuric acid and dicyclohexylcarbodiimide. In: Journal of the American Chemical Society. Band 91, Nr. 15, Juli 1969, S. 4273–4278, doi:10.1021/ja01043a041 (Herstellung durch Veresterung von Schwefelsäure).
- Everett E. Gilbert: The Reactions of Sulfur Trioxide, and Its Adducts, with Organic Compounds. In: Chemical Reviews. Band 62, Nr. 6, 1. Dezember 1962, S. 549–589, doi:10.1021/cr60220a003 (Synthese unter Verwendung von Schwefeltrioxid und Schwefeltrioxid-Komplexen).
- Hoe-Sup Byun, Linli He, Robert Bittman: Cyclic Sulfites and Cyclic Sulfates in Organic Synthesis. In: Tetrahedron. Band 56, Nr. 37, September 2000, S. 7051–7091, doi:10.1016/S0040-4020(00)00494-4 (Herstellung und Reaktionen von cyclischen Sulfaten).
- Constantina Papatriantafyllopoulou, Evy Manessi-Zoupa, Albert Escuer, Spyros P. Perlepes: The sulfate ligand as a promising “player” in 3d-metal cluster chemistry. In: Inorganica Chimica Acta. Band 362, Nr. 3, Februar 2009, S. 634–650, doi:10.1016/j.ica.2008.02.075 (Sulfat-Komplexe).
- Mineralogical Society of America, Paul H. Ribbe: Sulfate Minerals: Crystallography, Geochemistry, and Environmental Significance. Hrsg.: Charles N. Alpers, D. Nordstrom, John L. Jambor. Mineralogical Society of America, Washington, DC 2018, ISBN 978-1-5015-0866-0 (Sulfatminerale).
- Denis Barron, Luc Varin, Ragai K. Ibrahim, Jeffrey B. Harborne, Christine A. Williams: Sulphated flavonoids—an update. In: Phytochemistry. Band 27, Nr. 8, Januar 1988, S. 2375–2395, doi:10.1016/0031-9422(88)87003-1 (Naturstoffe, Flavonoidsulfate).
- Charles A. Strott: Sulfonation and Molecular Action. In: Endocrine Reviews. Band 23, Nr. 5, 1. Oktober 2002, S. 703–732, doi:10.1210/er.2001-0040 (Bedeutung von Sulfaten im menschlichen Körper).
- Ivica Blažević, Sabine Montaut, Franko Burčul, Carl Erik Olsen, Meike Burow, Patrick Rollin, Niels Agerbirk: Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. In: Phytochemistry. Band 169, Januar 2020, S. 112100 ff., doi:10.1016/j.phytochem.2019.112100 (Naturstoffe, Glucosinolate).
- Denis Barron, Luc Varin, Ragai K. Ibrahim, Jeffrey B. Harborne, Christine A. Williams: Sulphated flavonoids—an update. In: Phytochemistry. Band 27, Nr. 8, Januar 1988, S. 2375–2395, doi:10.1016/0031-9422(88)87003-1 (Naturstoffe, Flavonoidsulfate).
Weblinks
[Bearbeiten | Quelltext bearbeiten]- Literatur von und über Sulfate im Katalog der Deutschen Nationalbibliothek
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ Paolo Vitti: Mortars and masonry—structural lime and gypsum mortars in Antiquity and Middle Ages. In: Archaeological and Anthropological Sciences. Band 13, Nr. 10, Oktober 2021, doi:10.1007/s12520-021-01408-y.
- ↑ a b José da Silva Andrade Neto, Angeles G. De la Torre, Ana Paula Kirchheim: Effects of sulfates on the hydration of Portland cement – A review. In: Construction and Building Materials. Band 279, April 2021, S. 122428, doi:10.1016/j.conbuildmat.2021.122428.
- ↑ Zofia Anna Stos-Gale: Isotope archaeology: reading the past in metals, minerals, and bone. In: Endeavour. Band 16, Nr. 2, Juni 1992, S. 85–90, doi:10.1016/0160-9327(92)90007-C.
- ↑ W. Kloppmann, L. Leroux, P. Bromblet, P.-Y. Le Pogam, A. H. Cooper, N. Worley, C. Guerrot, A. T. Montech, A. M. Gallas, R. Aillaud: Competing English, Spanish, and French alabaster trade in Europe over five centuries as evidenced by isotope fingerprinting. In: Proceedings of the National Academy of Sciences. Band 114, Nr. 45, 7. November 2017, S. 11856–11860, doi:10.1073/pnas.1707450114, PMID 29078309, PMC 5692548 (freier Volltext).
- ↑ J. Ambers: Raman analysis of pigments from the Egyptian Old Kingdom. In: Journal of Raman Spectroscopy. Band 35, Nr. 8–9, August 2004, S. 768–773, doi:10.1002/jrs.1187.
- ↑ María Teresa Doménech-Carbó, María Luisa Vázquez de Agredos-Pascual, Laura Osete-Cortina, Antonio Doménech-Carbó, Núria Guasch-Ferré, Linda R. Manzanilla, Cristina Vidal-Lorenzo: Characterization of prehispanic cosmetics found in a burial of the ancient city of Teotihuacan (Mexico). In: Journal of Archaeological Science. Band 39, Nr. 4, April 2012, S. 1043–1062, doi:10.1016/j.jas.2011.12.001.
- ↑ Alana S. Lee, Peter J. Mahon, Dudley C. Creagh: Raman analysis of iron gall inks on parchment. In: Vibrational Spectroscopy. Band 41, Nr. 2, August 2006, S. 170–175, doi:10.1016/j.vibspec.2005.11.006.
- ↑ a b Karina Grömer, Gabriela Russ-Popa, Konstantina Saliari: Products of animal skin from Antiquity to the Medieval Period. In: Annalen des Naturhistorischen Museums in Wien. Serie A für Mineralogie und Petrographie, Geologie und Paläontologie, Anthropologie und Prähistorie. Band 119, 2017, S. 69–93, JSTOR:26342924.
- ↑ a b c d e Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 1. VCH, Weinheim 1985, ISBN 3-527-20101-7, S. 527–533 (Eintrag zu 'Aluminium Compounds, Inorganic').
- ↑ James C. Hill: Johann Glauber’s discovery of sodium sulfate – Sal Mirabile Glauberi. In: Journal of Chemical Education. Band 56, Nr. 9, September 1979, S. 593, doi:10.1021/ed056p593.
- ↑ Fortes, A. D.: "The Story of Epsomite." (PDF) Abgerufen am 13. März 2024.
- ↑ a b B. Szostakowski, P. Smitham, W.S. Khan: Plaster of Paris–Short History of Casting and Injured Limb Immobilzation. In: The Open Orthopaedics Journal. Band 11, Nr. 1, 17. April 2017, S. 291–296, doi:10.2174/1874325001711010291, PMID 28567158, PMC 5420179 (freier Volltext).
- ↑ a b Linda A. Hunter, Karen J. Gibbins: Magnesium Sulfate: Past, Present, and Future. In: Journal of Midwifery & Women’s Health. Band 56, Nr. 6, November 2011, S. 566–574, doi:10.1111/j.1542-2011.2011.00121.x.
- ↑ a b D.K Obatomi, P.H Bach: Biochemistry and Toxicology of the Diterpenoid Glycoside Atractyloside. In: Food and Chemical Toxicology. Band 36, Nr. 4, April 1998, S. 335–346, doi:10.1016/S0278-6915(98)00002-7.
- ↑ a b A Roy: Eugen Baumann and sulphate esters. In: Trends in Biochemical Sciences. Band 1, Nr. 10, Oktober 1976, S. N233–N234, doi:10.1016/0968-0004(76)90168-7.
- ↑ a b Ivica Blažević, Sabine Montaut, Franko Burčul, Carl Erik Olsen, Meike Burow, Patrick Rollin, Niels Agerbirk: Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. In: Phytochemistry. Band 169, Januar 2020, S. 112100, doi:10.1016/j.phytochem.2019.112100.
- ↑ a b Jeffrey B. Harborne: Flavonoid sulphates: A new class of sulphur compounds in higher plants. In: Phytochemistry. Band 14, Nr. 5-6, Juni 1975, S. 1147–1155, doi:10.1016/S0031-9422(00)98585-6.
- ↑ a b c d e Dallas L. Rabenstein: Heparin and heparan sulfate: structure and function. In: Natural Product Reports. Band 19, Nr. 3, 23. Mai 2002, S. 312–331, doi:10.1039/b100916h.
- ↑ J D Gregory, P W Robbins: METABOLISM OF SULFUR COMPOUNDS (SULFATE METABOLISM). In: Annual Review of Biochemistry. Band 29, Nr. 1, Juni 1960, S. 347–364, doi:10.1146/annurev.bi.29.070160.002023.
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 8. VCH, Weinheim/New York 1987, ISBN 3-527-20108-4, S. 493–494 (Eintrag zu 'Dialkyl Sulfates and Alkylsulfuric Acids').
- ↑ a b c d e f Everett E. Gilbert: The Reactions of Sulfur Trioxide, and Its Adducts, with Organic Compounds. In: Chemical Reviews. Band 62, Nr. 6, 1. Dezember 1962, S. 549–589, doi:10.1021/cr60220a003.
- ↑ a b c d Sulfate. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 10. März 2024.
- ↑ Vitriole. In: Spektrum.de, Lexikon der Geowissenschaften. Abgerufen am 3. März 2024.
- ↑ a b Eberhard Schweda, Gerhart Jander, Ewald Blasius: Jander/Blasius anorganische Chemie. 1: Theoretische Grundlagen und Qualitative Analyse. 19., völlig neu bearbeitete Auflage. Hirzel Verlag, Stuttgart 2022, ISBN 978-3-7776-3009-0, S. 247–248.
- ↑ Gmelins Handbuch der anorganischen Chemie: Radium, System Nummer 31. Achte Auflage, Verlag Chemie, Berlin 1927, S. 61–62.
- ↑ Eintrag zu Ammoniumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Natriumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Kaliumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Magnesiumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Calciumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Strontiumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Bariumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Aluminiumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Chrom(III)-sulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Mangan(II)-sulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Eisen(II)-sulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Nickel(II)-sulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Kupfer(II)-sulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Zinksulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Natriumhydrogensulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 14. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Kaliumhydrogensulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 14. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Kaliumaluminiumsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 14. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Chromalaun in der GESTIS-Stoffdatenbank des IFA, abgerufen am 14. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Dimethylsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Diethylsulfat in der GESTIS-Stoffdatenbank des IFA, abgerufen am 10. März 2024. (JavaScript erforderlich)
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5th ed Auflage. Band 8. VCH, Weinheim/New York 1987, ISBN 3-527-20108-4, S. 502–503 (Eintrag zu 'Dialkyl Sulfates and Alkylsulfuric Acids').
- ↑ Sulfite. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 25. März 2024.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.s06118.
- ↑ Sulfonsäuren. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 25. März 2024.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.s06117.
- ↑ Amidosulfonsäure. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 25. März 2024.
- ↑ Thiosulfate. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 25. März 2024.
- ↑ Dischwefelsäure. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 25. März 2024.
- ↑ Disulfate. In: Spektrum.de, Lexikon der Chemie. Abgerufen am 25. März 2024.
- ↑ Gilbert N. Lewis: The atom and the molecule. In: Journal of the American Chemical Society. Band 38, Nr. 4, April 1916, S. 762–785, doi:10.1021/ja02261a002.
- ↑ Laila Suidan, Jay K. Badenhoop, Eric D. Glendening, Frank Weinhold: Common Textbook and Teaching Misrepresentations of Lewis Structures. In: Journal of Chemical Education. Band 72, Nr. 7, Juli 1995, S. 583, doi:10.1021/ed072p583.
- ↑ William R. Cannon, B. Montgomery Pettitt, J. Andrew McCammon: Sulfate Anion in Water: Model Structural, Thermodynamic, and Dynamic Properties. In: The Journal of Physical Chemistry. Band 98, Nr. 24, Juni 1994, S. 6225–6230, doi:10.1021/j100075a027.
- ↑ Cory C. Pye, Wolfram W. Rudolph: An ab Initio and Raman Investigation of Sulfate Ion Hydration. In: The Journal of Physical Chemistry A. Band 105, Nr. 5, 1. Februar 2001, S. 905–912, doi:10.1021/jp003253n.
- ↑ S. John Louisnathan, Roderick J. Hill, G. V. Gibbs: Tetrahedral bond length variations in sulfates. In: Physics and Chemistry of Minerals. Band 1, Nr. 1, 1977, S. 53–69, doi:10.1007/BF00307979.
- ↑ Alexander V. Levanov, Oksana Ya. Isaikina, Ulkar D. Gurbanova, Valery V. Lunin: Dissociation Constants of Perchloric and Sulfuric Acids in Aqueous Solution. In: The Journal of Physical Chemistry B. Band 122, Nr. 23, 14. Juni 2018, S. 6277–6286, doi:10.1021/acs.jpcb.8b01947.
- ↑ Richard E. Lindstrom, Henry E. Wirth: Estimation of the bisulfate ion dissociation in solutions of sulfuric acid and sodium bisulfate. In: The Journal of Physical Chemistry. Band 73, Nr. 1, Januar 1969, S. 218–223, doi:10.1021/j100721a036.
- ↑ N. N. Greenwood, A. Earnshaw: Chemistry of the Elements. Elsevier Science, Burlington 2012, ISBN 978-0-08-050109-3, S. 711.
- ↑ Svetlana Malkhazova, Dmitry Orlov, Natalia Shartova, Sergey Starikov, Tatiana Puzanova: Healing Springs of Russia. Springer International Publishing, Cham 2022, ISBN 978-3-03083533-0, doi:10.1007/978-3-030-83534-7.
- ↑ A. Longinelli, H. Craig: Oxygen-18 Variations in Sulfate Ions in Sea Water and Saline Lakes. In: Science. Band 156, Nr. 3771, 7. April 1967, S. 56–59, doi:10.1126/science.156.3771.56.
- ↑ a b F J Millero: The Physical Chemistry of Seawater. In: Annual Review of Earth and Planetary Sciences. Band 2, Nr. 1, Mai 1974, S. 101–150, doi:10.1146/annurev.ea.02.050174.000533.
- ↑ a b William M. Last: Geolimnology of salt lakes. In: Geosciences Journal. Band 6, Nr. 4, Dezember 2002, S. 347–369, doi:10.1007/BF03020619.
- ↑ K. L. Falk, J. G. Tokuhisa, J. Gershenzon: The Effect of Sulfur Nutrition on Plant Glucosinolate Content: Physiology and Molecular Mechanisms. In: Plant Biology. Band 9, Nr. 5, September 2007, S. 573–581, doi:10.1055/s-2007-965431.
- ↑ a b c Shigeru Ōae, Joyce Takahashi Doi: Organic sulfur chemistry: structure and mechanism. CRC Press, Boca Raton, Fla 1991, ISBN 0-8493-4739-4, S. 360–363.
- ↑ Timothy H.J. Florin, Graeme Neale, Sara Goretski, John H Cummings: The Sulfate Content of Foods and Beverages. In: Journal of Food Composition and Analysis. Band 6, Nr. 2, Juni 1993, S. 140–151, doi:10.1006/jfca.1993.1016.
- ↑ a b c Sulfate. In: Spektrum.de, Lexikon der Geowissenschaften. Abgerufen am 10. März 2024.
- ↑ a b Mineralsystematik nach Strunz 9. Auflage von 2001 (incl. spätere Erweiterungen) – 7. Sulfate. In: Mineralienatlas Lexikon. Geolitho Stiftung, abgerufen am 12. März 2024.
- ↑ Ernest H. Nickel, Monte C. Nichols: IMA/CNMNC List of Minerals 2009. (PDF; 1,9 MB) In: cnmnc.units.it. IMA/CNMNC, Januar 2009, abgerufen am 12. März 2024 (englisch).
- ↑ a b Stefan Weiß: Das große Lapis Mineralienverzeichnis. Alle Mineralien von A – Z und ihre Eigenschaften. Stand 03/2018. 7., vollkommen neu bearbeitete und ergänzte Auflage. Weise, München 2018, ISBN 978-3-921656-83-9.
- ↑ Richard V. Gaines, H. Catherine W. Skinner, Eugene E. Foord, Brian Mason, Abraham Rosenzweig: Dana’s New Mineralogy. 8. Auflage. John Wiley & Sons, New York u. a. 1997, ISBN 0-471-19310-0.
- ↑ David Barthelmy: Dana’s New Sulfate Classification – 28 Sulfate Minerals. In: webmineral.com. Abgerufen am 12. März 2024 (englisch).
- ↑ a b c d e Manuel Bustillo Revuelta: Gypsum Products. In: Construction Materials. Springer International Publishing, Cham 2021, ISBN 978-3-03065206-7, S. 195–215, doi:10.1007/978-3-030-65207-4_8.
- ↑ Amit G. Reiss, Ittai Gavrieli, Yoav O. Rosenberg, Itay J. Reznik, Andreas Luttge, Simon Emmanuel, Jiwchar Ganor: Gypsum Precipitation under Saline Conditions: Thermodynamics, Kinetics, Morphology, and Size Distribution. In: Minerals. Band 11, Nr. 2, 30. Januar 2021, S. 141, doi:10.3390/min11020141.
- ↑ a b Niklas Wehmann, Christoph Lenting, Sandro Jahn: Calcium sulfates in planetary surface environments. In: Global and Planetary Change. Band 230, November 2023, S. 104257, doi:10.1016/j.gloplacha.2023.104257.
- ↑ R. C. Murray (2): Origin and Diagenesis of Gypsum and Anhydrite. In: SEPM Journal of Sedimentary Research. Vol. 34, 1964, doi:10.1306/74D710D2-2B21-11D7-8648000102C1865D.
- ↑ R. F. Parsons: Gypsophily in Plants – A Review. In: American Midland Naturalist. Band 96, Nr. 1, Juli 1976, S. 1, doi:10.2307/2424564.
- ↑ Amit G. Reiss, Ittai Gavrieli, Yoav O. Rosenberg, Itay J. Reznik, Andreas Luttge, Simon Emmanuel, Jiwchar Ganor: Gypsum Precipitation under Saline Conditions: Thermodynamics, Kinetics, Morphology, and Size Distribution. In: Minerals. Band 11, Nr. 2, 30. Januar 2021, S. 141, doi:10.3390/min11020141.
- ↑ Juan Manuel García-Ruiz, Roberto Villasuso, Carlos Ayora, Angels Canals, Fermín Otálora: Formation of natural gypsum megacrystals in Naica, Mexico. In: Geology. Band 35, Nr. 4, 2007, S. 327, doi:10.1130/G23393A.1.
- ↑ M. M. Al‐Kofahi, A. B. Hallak, H. A. Al‐Juwair, A. K. Saafin: Analysis of desert rose using PIXE and RBS techniques. In: X-Ray Spectrometry. Band 22, Nr. 1, Januar 1993, S. 23–27, doi:10.1002/xrs.1300220107.
- ↑ Robert D. Allen, Henry Kramer: Occurrence of bassanite in two desert basins in Southeastern California. In: American Mineralogist. Band 38, Nummer 11–12, 1953, S. 1266–1268.
- ↑ Delia-Georgeta Dumitraș et al.: Gypsum and bassanite in the bat guano deposit from the “dry” Cioclovina cave (Sureanu Mountains, Romania). In: Romanian Journal of Mineral Deposits. Band 81, 2004, S. 84–87.
- ↑ James J. Wray, Steven W. Squyres, Leah H. Roach, Janice L. Bishop, John F. Mustard, Eldar Z. Noe Dobrea: Identification of the Ca-sulfate bassanite in Mawrth Vallis, Mars. In: Icarus. Band 209, Nr. 2, Oktober 2010, S. 416–421, doi:10.1016/j.icarus.2010.06.001.
- ↑ Yves Langevin, François Poulet, Jean-Pierre Bibring, Brigitte Gondet: Sulfates in the North Polar Region of Mars Detected by OMEGA/Mars Express. In: Science. Band 307, Nr. 5715, 11. März 2005, S. 1584–1586, doi:10.1126/science.1109091.
- ↑ Martin Okrusch, Siegfried Matthes: Mineralogie: Eine Einführung in die spezielle Mineralogie, Petrologie und Lagerstättenkunde. 8. Auflage, Springer, Berlin / Heidelberg 2009, ISBN 978-3-540-78200-1, S. 107 ff.
- ↑ Harald G. Dill, Berthold Weber, Reiner Botz: Metalliferous duricrusts („orecretes“) – markers of weathering: A mineralogical and climatic-geomorphological approach to supergene Pb-Zn-Cu-Sb-P mineralization on different parent materials. In: Neues Jahrbuch für Mineralogie – Abhandlungen. Band 190, Nr. 2, 1. April 2013, S. 167, doi:10.1127/0077-7757/2013/0235.
- ↑ Ullmann’s encyclopedia of industrial chemistry. 7: Vol. A. Alphabetically arranged articles Chlorophenols to copper compounds. 5. Auflage. Band 7. VCH Verl.-Ges, Weinheim 1986, ISBN 3-527-20107-6, S. 577 (Eintrag zu 'Copper Compounds').
- ↑ N. V. Zubkova, I. V. Pekov, D. A. Ksenofontov, V. O. Yapaskurt, D. Yu. Pushcharovsky, E. G. Sidorov: Arcanite from Fumarole Exhalations of the Tolbachik Volcano (Kamchatka, Russia) and Its Crystal Structure. In: Doklady Earth Sciences. Band 479, Nr. 1, März 2018, S. 339–341, doi:10.1134/S1028334X18030121.
- ↑ Fabienne Bosselmann, Matthias Epple: Sulfate‐Containing Biominerals. In: Biomineralization. Wiley, 2008, ISBN 978-0-470-03525-2, S. 207–217, doi:10.1002/9780470986325.ch6.
- ↑ Crystallographic and morphological studies of the celestite skeleton of the acantharian species Phyllostaurus siculus. In: Proceedings of the Royal Society of London. Series B. Biological Sciences. Band 233, Nr. 1273, 23. Mai 1988, S. 393–405, doi:10.1098/rspb.1988.0029.
- ↑ John A. Raven, Andrew H. Knoll: Non-Skeletal Biomineralization by Eukaryotes: Matters of Moment and Gravity. In: Geomicrobiology Journal. Band 27, Nr. 6-7, 10. September 2010, S. 572–584, doi:10.1080/01490451003702990.
- ↑ a b Henry Tiemann, Ilka Sötje, Alexander Becker, Gerhard Jarms, Matthias Epple: Calcium sulfate hemihydrate (bassanite) statoliths in the cubozoan Carybdea sp. In: Zoologischer Anzeiger – A Journal of Comparative Zoology. Band 245, Nr. 1, Juli 2006, S. 13–17, doi:10.1016/j.jcz.2006.03.001.
- ↑ a b Henry Tiemann, Ilka Sötje, Gerhard Jarms, Carsten Paulmann, Matthias Epple, Bernd Hasse: Calcium sulfate hemihydrate in statoliths of deep-sea medusae. In: Journal of the Chemical Society, Dalton Transactions. Nr. 7, 26. März 2002, S. 1266–1268, doi:10.1039/b111524c.
- ↑ Fabienne Boßelmann, Matthias Epple, Ilka Sötje, Henry Tiemann: Statoliths of Calcium Sulfate Hemihydrate are used for Gravity Sensing in Rhopaliophoran Medusae (Cnidaria). In: Handbook of Biomineralization. Wiley, 2007, ISBN 978-3-527-31641-0, S. 261–272, doi:10.1002/9783527619443.ch15.
- ↑ T. Yoshimura, Y. Tamenori, H. Kawahata, A. Suzuki: Fluctuations of sulfate, S-bearing amino acids and magnesium in a giant clam shell. In: Biogeosciences. Band 11, Nr. 14, 24. Juli 2014, S. 3881–3886, doi:10.5194/bg-11-3881-2014.
- ↑ Natercia Barbosa, Jean-Michel Jaquet, Oscar Urquidi, Takuji B.M. Adachi, Montserrat Filella: Combined in vitro and in vivo investigation of barite microcrystals in Spirogyra (Zygnematophyceae, Charophyta). In: Journal of Plant Physiology. Band 276, September 2022, S. 153769, doi:10.1016/j.jplph.2022.153769.
- ↑ A. U. Dogan, M. Dogan, D. C. N. Chan, D. E. Wurster: Bassanite fromSalvadora persica: A new evaporitic biomineral. In: Carbonates and Evaporites. Band 20, Nr. 1, März 2005, S. 2–7, doi:10.1007/BF03175444.
- ↑ J. E. Penner, C. C. Chuang, K. Grant: Climate forcing by carbonaceous and sulfate aerosols. In: Climate Dynamics. Band 14, Nr. 12, 29. Oktober 1998, S. 839–851, doi:10.1007/s003820050259.
- ↑ O. Boucher, M. Pham: History of sulfate aerosol radiative forcings. In: Geophysical Research Letters. Band 29, Nr. 9, Mai 2002, doi:10.1029/2001GL014048.
- ↑ a b c Robert J. Charlson, Tom M. L. Wigley: Sulfate Aerosol and Climatic Change. In: Scientific American. Band 270, Nr. 2, Februar 1994, S. 48–57, doi:10.1038/scientificamerican0294-48.
- ↑ J. T. Kiehl, B. P. Briegleb: The Relative Roles of Sulfate Aerosols and Greenhouse Gases in Climate Forcing. In: Science. Band 260, Nr. 5106, 16. April 1993, S. 311–314, doi:10.1126/science.260.5106.311.
- ↑ Lauren R. Marshall, Elena C. Maters, Anja Schmidt, Claudia Timmreck, Alan Robock, Matthew Toohey: Volcanic effects on climate: recent advances and future avenues. In: Bulletin of Volcanology. Band 84, Nr. 5, Mai 2022, doi:10.1007/s00445-022-01559-3.
- ↑ Gene E. Likens, Richard F. Wright, James N. Galloway, Thomas J. Butler: Acid Rain. In: Scientific American. Band 241, Nr. 4, 1979, S. 43–51, JSTOR:24965312.
- ↑ M. Patrick McCormick, Larry W. Thomason, Charles R. Trepte: Atmospheric effects of the Mt Pinatubo eruption. In: Nature. Band 373, Nr. 6513, Februar 1995, S. 399–404, doi:10.1038/373399a0.
- ↑ Stefan Brönnimann, Daniel Krämer: Tambora und das «Jahr ohne Sommer» 1816. Klima, Mensch und Gesellschaft (= Geographica Bernensia. Band G90). Universität Bern, Bern 2016, ISBN 978-3-905835-45-8, DOI:10.4480/gb2016.g90.02.
- ↑ Matthew Toohey, Kirstin Krüger, Michael Sigl, Frode Stordal, Henrik Svensen: Climatic and societal impacts of a volcanic double event at the dawn of the Middle Ages. In: Climatic Change. Band 136, Nr. 3–4, Juni 2016, S. 401–412, doi:10.1007/s10584-016-1648-7.
- ↑ a b c d e f g h i j Charles A. Strott: Sulfonation and Molecular Action. In: Endocrine Reviews. Band 23, Nr. 5, 1. Oktober 2002, S. 703–732, doi:10.1210/er.2001-0040.
- ↑ a b c d e Eli Chapman, Michael D. Best, Sarah R. Hanson, Chi‐Huey Wong: Sulfotransferases: Structure, Mechanism, Biological Activity, Inhibition, and Synthetic Utility. In: Angewandte Chemie International Edition. Band 43, Nr. 27, 5. Juli 2004, S. 3526–3548, doi:10.1002/anie.200300631.
- ↑ Goran Malojčić, Robin L. Owen, Rudi Glockshuber: Structural and Mechanistic Insights into the PAPS-Independent Sulfotransfer Catalyzed by Bacterial Aryl Sulfotransferase and the Role of the DsbL/DsbI System in Its Folding. In: Biochemistry. Band 53, Nr. 11, 25. März 2014, S. 1870–1877, doi:10.1021/bi401725j.
- ↑ Nandita Shangari, Tom S. Chan, Peter J. O’Brien: Sulfation and Glucuronidation of Phenols: Implications in Coenyzme Q Metabolism. In: Methods in Enzymology. Band 400. Elsevier, 2005, ISBN 0-12-182805-0, S. 342–359, doi:10.1016/s0076-6879(05)00020-0.
- ↑ Vlasia Kastrinou Lampou, Birk Poller, Felix Huth, Audrey Fischer, Gerd A. Kullak-Ublick, Michael Arand, Heiko S. Schadt, Gian Camenisch: Novel insights into bile acid detoxification via CYP, UGT and SULT enzymes. In: Toxicology in Vitro. Band 87, März 2023, S. 105533, doi:10.1016/j.tiv.2022.105533.
- ↑ a b Paul W. Needs, Paul A. Kroon: Convenient syntheses of metabolically important quercetin glucuronides and sulfates. In: Tetrahedron. Band 62, Nr. 29, Juli 2006, S. 6862–6868, doi:10.1016/j.tet.2006.04.102.
- ↑ David W. Boulton, J. Paul Fawcett: The Pharmacokinetics of Levosalbutamol: What are the Clinical Implications? In: Clinical Pharmacokinetics. Band 40, Nr. 1, 2001, S. 23–40, doi:10.2165/00003088-200140010-00003.
- ↑ Sheldon Leong, Tammy Sirich: Indoxyl Sulfate—Review of Toxicity and Therapeutic Strategies. In: Toxins. Band 8, Nr. 12, 30. November 2016, S. 358, doi:10.3390/toxins8120358, PMID 27916890, PMC 5198552 (freier Volltext).
- ↑ a b c d Tadahisa Mikami, Hiroshi Kitagawa: Biosynthesis and function of chondroitin sulfate. In: Biochimica et Biophysica Acta (BBA) – General Subjects. Band 1830, Nr. 10, Oktober 2013, S. 4719–4733, doi:10.1016/j.bbagen.2013.06.006.
- ↑ H. Watanabe, Y. Yamada, K. Kimata: Roles of Aggrecan, a Large Chondroitin Sulfate Proteoglycan, in Cartilage Structure and Function. In: Journal of Biochemistry. Band 124, Nr. 4, 1. Oktober 1998, S. 687–693, doi:10.1093/oxfordjournals.jbchem.a022166.
- ↑ Nadeen O. Chahine, Faye H. Chen, Clark T. Hung, Gerard A. Ateshian: Direct Measurement of Osmotic Pressure of Glycosaminoglycan Solutions by Membrane Osmometry at Room Temperature. In: Biophysical Journal. Band 89, Nr. 3, September 2005, S. 1543–1550, doi:10.1529/biophysj.104.057315, PMID 15980166, PMC 1366659 (freier Volltext).
- ↑ Chiara Schiraldi, Donatella Cimini, Mario De Rosa: Production of chondroitin sulfate and chondroitin. In: Applied Microbiology and Biotechnology. Band 87, Nr. 4, Juli 2010, S. 1209–1220, doi:10.1007/s00253-010-2677-1.
- ↑ Vitor H. Pomin: Keratan sulfate: An up-to-date review. In: International Journal of Biological Macromolecules. Band 72, Januar 2015, S. 282–289, doi:10.1016/j.ijbiomac.2014.08.029.
- ↑ a b J. M. Trowbridge, R. L. Gallo: Dermatan sulfate: new functions from an old glycosaminoglycan. In: Glycobiology. Band 12, Nr. 9, 1. September 2002, S. 117R–125R, doi:10.1093/glycob/cwf066.
- ↑ Udo Häcker, Kent Nybakken, Norbert Perrimon: Heparan sulphate proteoglycans: the sweet side of development. In: Nature Reviews Molecular Cell Biology. Band 6, Nr. 7, Juli 2005, S. 530–541, doi:10.1038/nrm1681.
- ↑ a b Sultan N. Baytas, Robert J. Linhardt: Advances in the preparation and synthesis of heparin and related products. In: Drug Discovery Today. Band 25, Nr. 12, Dezember 2020, S. 2095–2109, doi:10.1016/j.drudis.2020.09.011, PMID 32947045, PMC 7718634 (freier Volltext).
- ↑ Yaping Chen, Terry Maguire, Ronald E. Hileman, Jonathan R. Fromm, Jeffrey D. Esko, Robert J. Linhardt, Rory M. Marks: Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. In: Nature Medicine. Band 3, Nr. 8, August 1997, S. 866–871, doi:10.1038/nm0897-866.
- ↑ a b Ger J. Strous, Jan Dekker: Mucin-Type Glycoproteins. In: Critical Reviews in Biochemistry and Molecular Biology. Band 27, Nr. 1-2, Januar 1992, S. 57–92, doi:10.3109/10409239209082559.
- ↑ Rama Bansil, Bradley S. Turner: Mucin structure, aggregation, physiological functions and biomedical applications. In: Current Opinion in Colloid & Interface Science. Band 11, Nr. 2-3, Juni 2006, S. 164–170, doi:10.1016/j.cocis.2005.11.001.
- ↑ Koichi Honke, Naoyuki Taniguchi: Sulfotransferases and sulfated oligosaccharides. In: Medicinal Research Reviews. Band 22, Nr. 6, November 2002, S. 637–654, doi:10.1002/med.10020.
- ↑ Matthias Eckhardt: The Role and Metabolism of Sulfatide in the Nervous System. In: Molecular Neurobiology. Band 37, Nr. 2-3, Juni 2008, S. 93–103, doi:10.1007/s12035-008-8022-3.
- ↑ W B Huttner: Tyrosine Sulfation and the Secretory Pathway. In: Annual Review of Physiology. Band 50, Nr. 1, Oktober 1988, S. 363–376, doi:10.1146/annurev.ph.50.030188.002051.
- ↑ a b Masahito Suiko, Ming-Cheh Liu: Change in binding affinities of 3Y1 secreted fibronectin upon desulfation of tyrosine-o-sulfate. In: Biochemical and Biophysical Research Communications. Band 154, Nr. 3, August 1988, S. 1094–1098, doi:10.1016/0006-291X(88)90253-7.
- ↑ Christof Niehrs, Roland Beißwanger, Wieland B. Huttner: Protein tyrosine sulfation, 1993 — an update. In: Chemico-Biological Interactions. Band 92, Nr. 1-3, Juni 1994, S. 257–271, doi:10.1016/0009-2797(94)90068-X.
- ↑ Evgeny Makogonenko, Galina Tsurupa, Kenneth Ingham, Leonid Medved: Interaction of Fibrin(ogen) with Fibronectin: Further Characterization and Localization of the Fibronectin-Binding Site. In: Biochemistry. Band 41, Nr. 25, 1. Juni 2002, S. 7907–7913, doi:10.1021/bi025770x.
- ↑ G. J. Bakus: Defensive mechanisms and ecology of some tropical holothurians. In: Marine Biology. Band 2, Nr. 1, Oktober 1968, S. 23–32, doi:10.1007/BF00351634.
- ↑ Vladimir I. Kalinin, Dmitry L. Aminin, Sergey A. Avilov, Alexandra S. Silchenko, Valentin A. Stonik: Triterpene Glycosides from Sea Cucucmbers (Holothuroidea, Echinodermata). Biological Activities and Functions. In: Studies in Natural Products Chemistry. Band 35. Elsevier, 2008, ISBN 978-0-444-53181-0, S. 135–196, doi:10.1016/s1572-5995(08)80006-3.
- ↑ S.L. Friess, R.C. Durant, J.D. Chanley, F.J. Fash: Role of the sulphate charge center in irreversible interactions of holothurin A with chemoreceptors. In: Biochemical Pharmacology. Band 16, Nr. 8, August 1967, S. 1617–1625, doi:10.1016/0006-2952(67)90140-2.
- ↑ J. D. Chanley, R. Ledeen, J. Wax, R. F. Nigrelli, Harry Sobotka: Holothurin. I. The Isolation, Properties and Sugar Components of Holothurin A 1. In: Journal of the American Chemical Society. Band 81, Nr. 19, Oktober 1959, S. 5180–5183, doi:10.1021/ja01528a040.
- ↑ V. I. Kalinin, V. A. Stonik: Glycosides of marine invertebrates. Structure of holothurin A2 from the holothurianHolothuria edulis. In: Chemistry of Natural Compounds. Band 18, Nr. 2, März 1982, S. 196–200, doi:10.1007/BF00577193.
- ↑ Ming‐Ping La, Cui Li, Ling Li, Peng Sun, Hua Tang, Bao‐Shu Liu, Wei Gong, Hua Han, Yang‐Hua Yi, Wen Zhang: New Bioactive Sulfated Alkenes from the Sea Cucumber Apostichopus japonicus. In: Chemistry & Biodiversity. Band 9, Nr. 6, Juni 2012, S. 1166–1171, doi:10.1002/cbdv.201100324.
- ↑ Anna Aiello, Sabina Carbonelli, Giuseppe Esposito, Ernesto Fattorusso, Teresa Iuvone, Marialuisa Menna: Novel Bioactive Sulfated Alkene and Alkanes from the Mediterranean Ascidian Halocynthia papillosa. In: Journal of Natural Products. Band 63, Nr. 11, November 2000, S. 1590–1592, doi:10.1021/np000281o.
- ↑ Susumu Ikegami, Yuji Kamiya, Saburo Tamura: A new steroidal sulfate obtained from a starfish saponin, asterosaponin a. In: Tetrahedron Letters. Band 14, Nr. 10, Januar 1973, S. 731–734, doi:10.1016/S0040-4039(01)95696-5.
- ↑ A. A. Kicha, N. V. Ivanchina, A. I. Kalinovsky, P. S. Dmitrenok, V. A. Stonik: Sulfated steroid compounds from the starfish Aphelasterias japonica of the Kuril population. In: Russian Chemical Bulletin. Band 50, Nr. 4, 2001, S. 724–727, doi:10.1023/A:1011337617808.
- ↑ Natalia V. Ivanchina, Alla A. Kicha, Timofey V. Malyarenko, Anatoly I. Kalinovsky, Pavel S. Dmitrenok, Valentin A. Stonik: Biosynthesis of polar steroids from the Far Eastern starfish Patiria (=Asterina) pectinifera. Cholesterol and cholesterol sulfate are converted into polyhydroxylated sterols and monoglycoside asterosaponin P1 in feeding experiments. In: Steroids. Band 78, Nr. 12–13, Dezember 2013, S. 1183–1191, doi:10.1016/j.steroids.2013.08.008.
- ↑ Yong Hae Kim, Yun Bong Kim, Mari Yotsu‐Yamashita: Potent Neurotoxins: Tetrodotoxin, Chiriquitoxin, and Zetekitoxin from Atelopus Frogs in Central America. In: Journal of Toxicology: Toxin Reviews. Band 22, Nr. 4, Januar 2003, S. 521–532, doi:10.1081/TXR-120026911.
- ↑ Mari Yotsu-Yamashita, Yong H. Kim, Samuel C. Dudley, Gaurav Choudhary, Arnold Pfahnl, Yasukatsu Oshima, John W. Daly: The structure of zetekitoxin AB, a saxitoxin analog from the Panamanian golden frog Atelopus zeteki: A potent sodium-channel blocker. In: Proceedings of the National Academy of Sciences. Band 101, Nr. 13, 30. März 2004, S. 4346–4351, doi:10.1073/pnas.0400368101, PMID 15070720, PMC 384749 (freier Volltext).
- ↑ a b c Ruslan Yatusevich, Sarah G. Mugford, Colette Matthewman, Tamara Gigolashvili, Henning Frerigmann, Sean Delaney, Anna Koprivova, Ulf-Ingo Flügge, Stanislav Kopriva: Genes of primary sulfate assimilation are part of the glucosinolate biosynthetic network in Arabidopsis thaliana: Glucosinolate biosynthesis and sulfate assimilation. In: The Plant Journal. Band 62, Nr. 1, April 2010, S. 1–11, doi:10.1111/j.1365-313X.2009.04118.x.
- ↑ a b c Don Brian Clarke: Glucosinolates, structures and analysis in food. In: Analytical Methods. Band 2, Nr. 4, 2010, S. 310, doi:10.1039/b9ay00280d.
- ↑ a b c d e Barbara Ann Halkier, Jonathan Gershenzon: Biology and Biochemistry of Glucosinolates. In: Annual Review of Plant Biology. Band 57, Nr. 1, 1. Juni 2006, S. 303–333, doi:10.1146/annurev.arplant.57.032905.105228.
- ↑ Fekadu Kassie, Brenda Laky, Richard Gminski, Volker Mersch-Sundermann, Gerlinde Scharf, Evelyn Lhoste, Siegfried Kansmüller: Effects of garden and water cress juices and their constituents, benzyl and phenethyl isothiocyanates, towards benzo(a)pyrene-induced DNA damage: a model study with the single cell gel electrophoresis/Hep G2 assay. In: Chemico-Biological Interactions. Band 142, Nr. 3, Januar 2003, S. 285–296, doi:10.1016/S0009-2797(02)00123-0.
- ↑ a b c d e Niels Agerbirk, Carl Erik Olsen: Glucosinolate structures in evolution. In: Phytochemistry. Band 77, Mai 2012, S. 16–45, doi:10.1016/j.phytochem.2012.02.005.
- ↑ K. L. Falk, J. G. Tokuhisa, J. Gershenzon: The Effect of Sulfur Nutrition on Plant Glucosinolate Content: Physiology and Molecular Mechanisms. In: Plant Biology. Band 9, Nr. 5, September 2007, S. 573–581, doi:10.1055/s-2007-965431.
- ↑ a b c d e Andreas Ratzka, Heiko Vogel, Daniel J. Kliebenstein, Thomas Mitchell-Olds, Juergen Kroymann: Disarming the mustard oil bomb. In: Proceedings of the National Academy of Sciences. Band 99, Nr. 17, 20. August 2002, S. 11223–11228, doi:10.1073/pnas.172112899, PMID 12161563, PMC 123237 (freier Volltext).
- ↑ a b Denis Barron, Luc Varin, Ragai K. Ibrahim, Jeffrey B. Harborne, Christine A. Williams: Sulphated flavonoids—an update. In: Phytochemistry. Band 27, Nr. 8, Januar 1988, S. 2375–2395, doi:10.1016/0031-9422(88)87003-1.
- ↑ Abdelali Hannoufa, Luc Varin, Ragai K. Ibrahim: Spatial Distribution of Flavonoid Conjugates in Relation to Glucosyltransferase and Sulfotransferase Activities in Flaveria bidentis. In: Plant Physiology. Band 97, Nr. 1, 1. September 1991, S. 259–263, doi:10.1104/pp.97.1.259, PMID 16668379, PMC 1080992 (freier Volltext).
- ↑ Luc Varin, Ragai K. Ibrahim: Partial Purification and Characterization of Three Flavonol-Specific Sulfotransferases from Flaveria chloraefolia. In: Plant Physiology. Band 90, Nr. 3, 1. Juli 1989, S. 977–981, doi:10.1104/pp.90.3.977, PMID 16666908, PMC 1061831 (freier Volltext).
- ↑ Hui Yang, Petr Protiva, Baoliang Cui, Cuiying Ma, Scott Baggett, Vanessa Hequet, Scott Mori, I. Bernard Weinstein, Edward J. Kennelly: New Bioactive Polyphenols from Theobroma g randiflorum (“Cupuaçu”). In: Journal of Natural Products. Band 66, Nr. 11, Dezember 2003, S. 1501–1504, doi:10.1021/np034002j.
- ↑ Quan-Xiang Wu, Xiao-Feng He, Chun-Xiao Jiang, Wei Zhang, Zhuan-Ning Shi, Hong-Fang Li, Ying Zhu: Two novel bioactive sulfated guaiane sesquiterpenoid salt alkaloids from the aerial parts of Scorzonera divaricata. In: Fitoterapia. Band 124, Januar 2018, S. 113–119, doi:10.1016/j.fitote.2017.10.011.
- ↑ Reuben A. Sessa, Mark H. Bennett, Mervyn J. Lewis, John W. Mansfield, Michael H. Beale: Metabolite Profiling of Sesquiterpene Lactones from Lactuca Species. In: Journal of Biological Chemistry. Band 275, Nr. 35, September 2000, S. 26877–26884, doi:10.1016/S0021-9258(19)61456-0.
- ↑ Filippo Imperato: Sulphate esters of hydroxycinnamic acid—sugar derivatives from Adiantum capillus-veneris. In: Phytochemistry. Band 21, Nr. 11, Januar 1982, S. 2717–2718, doi:10.1016/0031-9422(82)83105-1.
- ↑ Toru Ishikawa, Kyoko Kondo, Junichi Kitajima: Water-Soluble Constituents of Coriander. In: Chemical and Pharmaceutical Bulletin. Band 51, Nr. 1, 2003, S. 32–39, doi:10.1248/cpb.51.32.
- ↑ Antonin Chevenier, Diane Jouanneau, Elizabeth Ficko-Blean: Carrageenan biosynthesis in red algae: A review. In: The Cell Surface. Band 9, Dezember 2023, S. 100097, doi:10.1016/j.tcsw.2023.100097, PMID 37396716, PMC 10311240 (freier Volltext).
- ↑ John V. Mulcahy, James R. Walker, Jeffrey E. Merit, Alan Whitehead, J. Du Bois: Synthesis of the Paralytic Shellfish Poisons (+)-Gonyautoxin 2, (+)-Gonyautoxin 3, and (+)-11,11-Dihydroxysaxitoxin. In: Journal of the American Chemical Society. Band 138, Nr. 18, 11. Mai 2016, S. 5994–6001, doi:10.1021/jacs.6b02343.
- ↑ Masayuki Satake, Lincoln MacKenzie, Takeshi Yasumoto: Identification of Protoceratium reticulatum as the biogenetic origin of yessotoxin. In: Natural Toxins. Band 5, Nr. 4, Juli 1997, S. 164–167, doi:10.1002/19970504NT7.
- ↑ Lesley Rhodes, Paul McNabb, Miguel de Salas, Lyn Briggs, Veronica Beuzenberg, Melissa Gladstone: Yessotoxin production by Gonyaulax spinifera. In: Harmful Algae. Band 5, Nr. 2, März 2006, S. 148–155, doi:10.1016/j.hal.2005.06.008.
- ↑ K. C. Nicolaou, Kevin P. Cole, Michael O. Frederick, Robert J. Aversa, Ross M. Denton: Chemical Synthesis of the GHIJK Ring System and Further Experimental Support for the Originally Assigned Structure of Maitotoxin. In: Angewandte Chemie International Edition. Band 46, Nr. 46, 26. November 2007, S. 8875–8879, doi:10.1002/anie.200703742.
- ↑ Michio Murata, Hideo Naoki, Takashi Iwashita, Shigeki Matsunaga, Masahiro Sasaki, Akihiro Yokoyama, Takeshi Yasumoto: Structure of maitotoxin. In: Journal of the American Chemical Society. Band 115, Nr. 5, März 1993, S. 2060–2062, doi:10.1021/ja00058a075.
- ↑ David L. Burgoyne, Thomas K. Hemscheidt, Richard E. Moore, Maria T. C. Runnegar: Biosynthesis of Cylindrospermopsin. In: The Journal of Organic Chemistry. Band 65, Nr. 1, 1. Januar 2000, S. 152–156, doi:10.1021/jo991257m.
- ↑ Rong Yue, Meng Li, Yue Wang, Ying Guan, Jing Zhang, Zhongli Yan, Fufeng Liu, Fuping Lu, Huitu Zhang: Insight into enzyme-catalyzed aziridine formation mechanism in ficellomycin biosynthesis. In: European Journal of Medicinal Chemistry. Band 204, Oktober 2020, S. 112639, doi:10.1016/j.ejmech.2020.112639.
- ↑ Cornelius G. Friedrich: Physiology and Genetics of Sulfur-oxidizing Bacteria. In: Advances in Microbial Physiology. Band 39. Elsevier, 1997, ISBN 0-12-027739-5, S. 235–289, doi:10.1016/s0065-2911(08)60018-1.
- ↑ Daniel C. Brune: Sulfur oxidation by phototrophic bacteria. In: Biochimica et Biophysica Acta (BBA) – Bioenergetics. Band 975, Nr. 2, Juli 1989, S. 189–221, doi:10.1016/S0005-2728(89)80251-8.
- ↑ Harald G. Dill, Berthold Weber, Reiner Botz: Metalliferous duricrusts („orecretes“) – markers of weathering: A mineralogical and climatic-geomorphological approach to supergene Pb-Zn-Cu-Sb-P mineralization on different parent materials. In: Neues Jahrbuch für Mineralogie – Abhandlungen. Band 190, Nr. 2, 1. April 2013, S. 155, doi:10.1127/0077-7757/2013/0235.
- ↑ David A. Stahl, Susan Fishbain, Michael Klein, Brett J. Baker, Michael Wagner: Origins and diversification of sulfate-respiring microorganisms. Hrsg.: Antonie van Leeuwenhoek. Band 81, Nr. 1/4, 2002, S. 189–195, doi:10.1023/A:1020506415921.
- ↑ Julie Leloup, Alexander Loy, Nina J. Knab, Christian Borowski, Michael Wagner, Bo Barker Jørgensen: Diversity and abundance of sulfate‐reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea. In: Environmental Microbiology. Band 9, Nr. 1, Januar 2007, S. 131–142, doi:10.1111/j.1462-2920.2006.01122.x.
- ↑ Kasper Urup Kjeldsen, Alexander Loy, Trine Fredlund Jakobsen, Trine Rolighed Thomsen, Michael Wagner, Kjeld Ingvorsen: Diversity of sulfate-reducing bacteria from an extreme hypersaline sediment, Great Salt Lake (Utah): Diversity of SRB in Great Salt Lake. In: FEMS Microbiology Ecology. Band 60, Nr. 2, Mai 2007, S. 287–298, doi:10.1111/j.1574-6941.2007.00288.x.
- ↑ Trine Fredlund Jakobsen, Kasper Urup Kjeldsen, Kjeld Ingvorsen: Desulfohalobium utahense sp. nov., a moderately halophilic, sulfate-reducing bacterium isolated from Great Salt Lake. In: International Journal of Systematic and Evolutionary Microbiology. Band 56, Nr. 9, 1. September 2006, S. 2063–2069, doi:10.1099/ijs.0.64323-0.
- ↑ Pascal Philippot, Mark Van Zuilen, Kevin Lepot, Christophe Thomazo, James Farquhar, Martin J. Van Kranendonk: Early Archaean Microorganisms Preferred Elemental Sulfur, Not Sulfate. In: Science. Band 317, Nr. 5844, 14. September 2007, S. 1534–1537, doi:10.1126/science.1145861.
- ↑ G R Gibson, G T Macfarlane, J H Cummings: Sulphate reducing bacteria and hydrogen metabolism in the human large intestine. In: Gut. Band 34, Nr. 4, 1. April 1993, S. 437–439, doi:10.1136/gut.34.4.437.
- ↑ a b c Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 4. VCH, Weinheim 1985, ISBN 3-527-20104-1, S. 560–562 (Eintrag zu 'Calcium Sulfate').
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 3. VCH, Weinheim 1985, ISBN 3-527-20103-3, S. 328 (Eintrag zu 'Barium and Barium Compounds').
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 15. VCH, Weinheim/New York 1990, ISBN 3-527-20115-7, S. 620 (Eintrag zu 'Magnesium Compounds').
- ↑ Ullmann’s encyclopedia of industrial chemistry. 7: Vol. A. Alphabetically arranged articles Chlorophenols to copper compounds. 5. Auflage. Band 7. VCH Verl.-Ges, Weinheim 1986, ISBN 3-527-20107-6, S. 81 (Eintrag zu 'Chromium Compounds').
- ↑ Ullmann’s encyclopedia of industrial chemistry: Vol. A. Alphabetically arranged articles Chlorophenols to copper compounds. 5. Auflage. Band 7. VCH Verl.-Ges, Weinheim 1986, ISBN 3-527-20107-6, S. 578 (Eintrag zu 'Copper Compounds').
- ↑ Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 14. VCH, Weinheim/New York 1989, ISBN 3-527-20114-9, S. 591–592 (Eintrag zu 'Iron Compounds').
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 15. VCH, Weinheim/New York 1990, ISBN 3-527-20115-7, S. 410 (Eintrag zu 'Lithium and Lithium Compounds').
- ↑ a b c d Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 16. VCH, Weinheim/New York 1990, ISBN 3-527-20116-5, S. 132–133 (Eintrag zu 'Manganese Compounds').
- ↑ a b c d Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 17. VCH, Weinheim/New York 1991, ISBN 3-527-20117-3, S. 238 (Eintrag zu 'Nickel Compounds').
- ↑ a b c Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 22. VCH, Weinheim 1993, ISBN 3-527-20122-X, S. 84–91 (Eintrag zu 'Potassium Compounds').
- ↑ Tomi Gominšek, Andrej Lubej, Ciril Pohar: Continuous precipitation of calcium sulfate dihydrate from waste sulfuric acid and lime. In: Journal of Chemical Technology & Biotechnology. Band 80, Nr. 8, August 2005, S. 939–947, doi:10.1002/jctb.1266.
- ↑ Zhilong Zhu, You Peng, T. Alan Hatton, Kamal Samrane, Allan S. Myerson, Richard D. Braatz: Crystallization of Calcium Sulphate During Phosphoric Acid Production: Modeling Particle Shape and Size Distribution. In: Procedia Engineering. Band 138, 2016, S. 390–402, doi:10.1016/j.proeng.2016.02.098.
- ↑ a b c d e f g Rami A. Al-Horani, Umesh R. Desai: Chemical sulfation of small molecules—advances and challenges. In: Tetrahedron. Band 66, Nr. 16, April 2010, S. 2907–2918, doi:10.1016/j.tet.2010.02.015, PMID 20689724, PMC 2913517 (freier Volltext).
- ↑ Ryo Takano, Shuichi Yoshikawa, Takashi Ueda, Kaeko Hayashi, Susumu Hirase, Saburo Hara: Sulfation of Polysaccharides with Sulfuric Acid Mediated by Dicyclohexylcarbodiimide. In: Journal of Carbohydrate Chemistry. Band 15, Nr. 4, Mai 1996, S. 449–457, doi:10.1080/07328309608005665.
- ↑ Charles P. Hoiberg, Ralph O. Mumma: Selective hydroxyl sulfation by a dicyclohexylcarbodiimide-mediated reaction. In: Biochimica et Biophysica Acta (BBA) – General Subjects. Band 177, Nr. 1, Februar 1969, S. 149–151, doi:10.1016/0304-4165(69)90075-0.
- ↑ Charles P. Hoiberg, Ralph O. Mumma: Preparation of sulfate esters. Reactions of various alcohols, phenols, amines, mercaptans, and oximes with sulfuric acid and dicyclohexylcarbodiimide. In: Journal of the American Chemical Society. Band 91, Nr. 15, Juli 1969, S. 4273–4278, doi:10.1021/ja01043a041.
- ↑ H. G. Harris, D. M. Himmelblau: Kinetics of the Reaction of Ethylene with Sulfuric Acid. In: Journal of Chemical & Engineering Data. Band 9, Nr. 1, 1. Januar 1964, S. 61–65, doi:10.1021/je60020a018.
- ↑ Swades Kumar Chaudhuri, Man Mohan Sharma: Absorption of ethylene in concentrated sulfuric acid. In: Industrial & Engineering Chemistry Research. Band 30, Nr. 2, Februar 1991, S. 339–345, doi:10.1021/ie00050a010.
- ↑ Lyle F. Albright, Mark A. Spalding, James A. Nowinski, Robert M. Ybarra, Roger E. Eckert: Alkylation of isobutane with C4 olefins. 1. First-step reactions using sulfuric acid catalyst. In: Industrial & Engineering Chemistry Research. Band 27, Nr. 3, März 1988, S. 381–386, doi:10.1021/ie00075a003.
- ↑ Zhikui He, Honghong Liu, Shanshan Gui, Huai Liu, Jianyuan Yang, Qigen Guo, Xiangrong Ye, Binghuo Zhang: Procoagulant substances and mechanisms of hemostatic herb Eclipta alba. In: Process Biochemistry. Band 122, November 2022, S. 103–114, doi:10.1016/j.procbio.2022.08.027.
- ↑ Denis Barron, Ragal K. Ibrahim: Synthesis of flavonoid sulfates: 1. stepwise sulfation of positions 3, 7, and 4 using N,N'-dicyclohexylcarbodiimide and tetrabutylammonium hydrogen sulfate. In: Tetrahedron. Band 43, Nr. 22, Januar 1987, S. 5197–5202, doi:10.1016/S0040-4020(01)87695-X.
- ↑ a b c Marta Correia-da-Silva, Emília Sousa, Madalena M. M. Pinto: Emerging Sulfated Flavonoids and other Polyphenols as Drugs: Nature as an Inspiration: EMERGING SULFATED POLYPHENOLS AS DRUGS. In: Medicinal Research Reviews. Band 34, Nr. 2, März 2014, S. 223–279, doi:10.1002/med.21282.
- ↑ Martin Cupery: SULFAMIC ACID A NEW INDUSTRIAL CHEMICAL. In: Industrial & Engineering Chemistry. Band 30, Nr. 6, Juni 1938, S. 627–631, doi:10.1021/ie50342a005.
- ↑ Aleksandr S. Kazachenko, Yuriy N. Malyar, Natalya Yu. Vasilyeva, Valentina S. Borovkova, Noureddine Issaoui: Optimization of guar gum galactomannan sulfation process with sulfamic acid. In: Biomass Conversion and Biorefinery. Band 13, Nr. 11, Juli 2023, S. 10041–10050, doi:10.1007/s13399-021-01895-y.
- ↑ Organic chemistry of sulfur. Plenum Press, New York 1977, ISBN 0-306-30740-5, S. 656.
- ↑ a b c d Hoe-Sup Byun, Linli He, Robert Bittman: Cyclic Sulfites and Cyclic Sulfates in Organic Synthesis. In: Tetrahedron. Band 56, Nr. 37, September 2000, S. 7051–7091, doi:10.1016/S0040-4020(00)00494-4.
- ↑ a b c Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 8. VCH, Weinheim/New York 1987, ISBN 3-527-20108-4, S. 495–497 (Eintrag zu 'Dialkyl Sulfates and Alkylsulfuric Acids').
- ↑ Shuaishuai Yue, Guoping Ding, Ye Zheng, Chunlan Song, Peng Xu, Biao Yu, Jiakun Li: Dimethyl sulfate and diisopropyl sulfate as practical and versatile O-sulfation reagents. In: Nature Communications. Band 15, Nr. 1, 29. Februar 2024, doi:10.1038/s41467-024-46214-x, PMID 38424087, PMC 10904734 (freier Volltext).
- ↑ J.P. Dusza, J.P. Joseph, Seymour Bernstein: Steroid conjugates IV. The preparation of steroid sulfates with triethylamine-sulfur trioxide. In: Steroids. Band 12, Nr. 1, Juli 1968, S. 49–61, doi:10.1016/S0039-128X(68)80079-0.
- ↑ Donald J.L. Jones, Rebekah Jukes-Jones, Richard D. Verschoyle, Peter B. Farmer, Andreas Gescher: A synthetic approach to the generation of quercetin sulfates and the detection of quercetin 3′-O-sulfate as a urinary metabolite in the rat. In: Bioorganic & Medicinal Chemistry. Band 13, Nr. 24, Dezember 2005, S. 6727–6731, doi:10.1016/j.bmc.2005.07.021.
- ↑ Shiroh Futaki, Takashi Taike, Takeshi Yagami, Toyoko Ogawa, Tadashi Akita, Kouki Kitagawa: Use of dimethylformamide–sulphur trioxide complex as a sulphating agent of tyrosine. In: J. Chem. Soc., Perkin Trans. 1. Nr. 6, 1990, S. 1739–1744, doi:10.1039/P19900001739.
- ↑ Socorro Vázquez Campos, Les P. Miranda, Morten Meldal: Preparation of novel O-sulfated amino acid building blocks with improved acid stability for Fmoc-based solid-phase peptide synthesis. In: Journal of the Chemical Society, Perkin Transactions 1. Nr. 5, 22. Februar 2002, S. 682–686, doi:10.1039/b107320f.
- ↑ Arjun Raghuraman, Muhammad Riaz, Michael Hindle, Umesh R. Desai: Rapid and efficient microwave-assisted synthesis of highly sulfated organic scaffolds. In: Tetrahedron Letters. Band 48, Nr. 38, September 2007, S. 6754–6758, doi:10.1016/j.tetlet.2007.07.100, PMID 18797498, PMC 2084256 (freier Volltext).
- ↑ Suresh M. Sethna: The Elbs Persulfate Oxidation. In: Chemical Reviews. Band 49, Nr. 1, 1. August 1951, S. 91–101, doi:10.1021/cr60152a002.
- ↑ Xiao Gang, Yimin Zhu, Henning Birch, Hans Aage Hjuler, Niels J. Bjerrum: Iodine as catalyst for the direct oxidation of methane to methyl sulfates in oleum. In: Applied Catalysis A: General. Band 261, Nr. 1, April 2004, S. 91–98, doi:10.1016/j.apcata.2003.10.039.
- ↑ a b Iván Ayuso‐Fernández, Miquel A. Galmés, Agatha Bastida, Eduardo García‐Junceda: Aryl Sulfotransferase from Haliangium ochraceum: A Versatile Tool for the Sulfation of Small Molecules. In: ChemCatChem. Band 6, Nr. 4, April 2014, S. 1059–1065, doi:10.1002/cctc.201300853.
- ↑ a b Michael A. van der Horst, Johan F. T. van Lieshout, Aleksandra Bury, Aloysius F. Hartog, Ron Wever: Sulfation of Various Alcoholic Groups by an Arylsulfate Sulfotransferase from Desulfitobacterium hafniense and Synthesis of Estradiol Sulfate. In: Advanced Synthesis & Catalysis. Band 354, Nr. 18, 14. Dezember 2012, S. 3501–3508, doi:10.1002/adsc.201200564.
- ↑ Vadim V. Mozhaev, Yuri L. Khmelnitsky, Fernando Sanchez‐Riera, Julie Maurina‐Brunker, Reinhardt A. Rosson, Alan D. Grund: Arylsulfotransferase from Clostridium innocuum —A new enzyme catalyst for sulfation of phenol‐containing compounds. In: Biotechnology and Bioengineering. Band 78, Nr. 5, 5. Juni 2002, S. 567–575, doi:10.1002/bit.10229.
- ↑ Aloysius F. Hartog, Ron Wever: Sulfation made easy: A new versatile donor for enzymatic sulfation by a bacterial arylsulfotransferase. In: Journal of Molecular Catalysis B: Enzymatic. Band 129, Juli 2016, S. 43–46, doi:10.1016/j.molcatb.2016.04.007.
- ↑ Hiroaki Tagawa: Thermal decomposition temperatures of metal sulfates. In: Thermochimica Acta. Band 80, Nr. 1, Oktober 1984, S. 23–33, doi:10.1016/0040-6031(84)87181-6.
- ↑ Chunbin Guo, Jingjing Zou, Yinshan Jiang, Tianping Huang, Yan Cheng, Cundi Wei: Thermal activation of coal fly ash by sodium hydrogen sulfate for alumina extraction. In: Journal of Materials Science. Band 49, Nr. 12, Juni 2014, S. 4315–4322, doi:10.1007/s10853-014-8127-1.
- ↑ a b Rasmus Fehrmann, Niels Holger Hansen, Niels J. Bjerrum: Raman spectroscopic and spectrophotometric study of the system potassium pyrosulfate-potassium hydrogen sulfate in the temperature range 200-450.degree.C. In: Inorganic Chemistry. Band 22, Nr. 26, Dezember 1983, S. 4009–4014, doi:10.1021/ic00168a038.
- ↑ Fathi Habashi, Shaheer A. Mikhail, Kim Vo Van: Reduction of sulfates by hydrogen. In: Canadian Journal of Chemistry. Band 54, Nr. 23, 1. Dezember 1976, S. 3646–3650, doi:10.1139/v76-524.
- ↑ John H. Cameron, Thomas M. Grace: Kinetic study of sulfate reduction with carbon. In: Industrial & Engineering Chemistry Fundamentals. Band 22, Nr. 4, November 1983, S. 486–494, doi:10.1021/i100012a021.
- ↑ Sui-Qin Yang, Yu-Hong Cui, Ya-Yue Liu, Zheng-Qian Liu, Xue-Yan Li: Electrochemical generation of persulfate and its performance on 4-bromophenol treatment. In: Separation and Purification Technology. Band 207, Dezember 2018, S. 461–469, doi:10.1016/j.seppur.2018.06.071.
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 8. VCH, Weinheim/New York 1987, ISBN 3-527-20108-4, S. 499–502 (Eintrag zu 'Dialkyl Sulfates and Alkylsulfuric Acids').
- ↑ Barry Martin Trost, Ian Flemming, Steven V. Ley: Comprehensive organic synthesis: selectivity, strategy & efficiency in modern organic chemistry. Band 6. Pergamon press, Oxford/New York/Seoul 1991, ISBN 0-08-035929-9, S. 206.
- ↑ Frank H. Stodola: Base-Catalyzed Preparation of Methyl and Ethyl Esters of Carboxylic Acids. In: The Journal of Organic Chemistry. Band 29, Nr. 8, August 1964, S. 2490–2491, doi:10.1021/jo01031a535.
- ↑ Asit K. Chakraborti, Basak, Vikas Grover: Chemoselective Protection of Carboxylic Acid as Methyl Ester: A Practical Alternative to Diazomethane Protocol. In: The Journal of Organic Chemistry. Band 64, Nr. 21, 1. Oktober 1999, S. 8014–8017, doi:10.1021/jo990035l.
- ↑ Maurizio Selva, Alvise Perosa: Green chemistry metrics: a comparative evaluation of dimethyl carbonate, methyl iodide, dimethyl sulfate and methanol as methylating agents. In: Green Chemistry. Band 10, Nr. 4, 2008, S. 457, doi:10.1039/b713985c.
- ↑ Henry Gilman, Nathaniel J. Beaber: THE ALKYLATION OF MERCAPTANS BY MEANS OF SULFONIC ESTERS. In: Journal of the American Chemical Society. Band 47, Nr. 5, Mai 1925, S. 1449–1451, doi:10.1021/ja01682a034.
- ↑ Howard L. Kane, Alexander Lowy: Reactions of Alkyl Sulfates, Tetraethyl Orthosilicate and Diethyl Carbonate in Friedel-Crafts Syntheses 1,2. In: Journal of the American Chemical Society. Band 58, Nr. 12, Dezember 1936, S. 2605–2608, doi:10.1021/ja01303a064.
- ↑ Comprehensive organic synthesis: selectivity, strategy, and efficiency in modern organic chemistry. Band 6. Pergamon Press, Oxford/New York 1991, ISBN 0-08-035929-9, S. 503.
- ↑ a b Comprehensive organic synthesis: selectivity, strategy & efficiency in modern organic chemistry. Band 7. Pergamon Press, Oxford 1991, ISBN 0-08-035929-9, S. 431–432.
- ↑ E. T. Kaiser, Manuel. Panar, F. H. Westheimer: The Hydrolysis of Some Cyclic Esters of Sulfuric Acid. In: Journal of the American Chemical Society. Band 85, Nr. 5, März 1963, S. 602–607, doi:10.1021/ja00888a028.
- ↑ Mayer B. Goren, Mary E. Kochansky: Stringent requirement for electrophiles in the facile solvolytic hydrolysis of neutral sulfate ester salts. In: The Journal of Organic Chemistry. Band 38, Nr. 20, Oktober 1973, S. 3510–3513, doi:10.1021/jo00960a016.
- ↑ Organic chemistry of sulfur. Plenum Press, New York 1977, ISBN 0-306-30740-5, S. 658.
- ↑ Constantina Papatriantafyllopoulou, Evy Manessi-Zoupa, Albert Escuer, Spyros P. Perlepes: The sulfate ligand as a promising “player” in 3d-metal cluster chemistry. In: Inorganica Chimica Acta. Band 362, Nr. 3, Februar 2009, S. 634–650, doi:10.1016/j.ica.2008.02.075.
- ↑ Dominik Schaniel, Theo Woike, Norwid-R. Behrnd, Jürg Hauser, Karl W. Krämer, Teodora Todorova, Bernard Delley: Photogeneration of Nitrosyl Linkage Isomers in Octahedrally Coordinated Platinum Complexes in the Red Spectral Range. In: Inorganic Chemistry. Band 48, Nr. 23, 7. Dezember 2009, S. 11399–11406, doi:10.1021/ic901392q.
- ↑ R. W. Horn, Edward Weissberger, James P. Collman: Oxygen-18 study of the reaction between iridium- and platinum-oxygen complexes and sulfur dioxide to form coordinated sulfate. In: Inorganic Chemistry. Band 9, Nr. 10, Oktober 1970, S. 2367–2371, doi:10.1021/ic50092a036.
- ↑ Christopher David. Cook, G. S. Jauhal: Oxidation of coordinated ligands. Sulfato and nitrato complexes of platinum. In: Journal of the American Chemical Society. Band 89, Nr. 12, Juni 1967, S. 3066–3067, doi:10.1021/ja00988a057.
- ↑ Christoph Hennig, Atsushi Ikeda-Ohno, Satoru Tsushima, Andreas C. Scheinost: The Sulfate Coordination of Np(IV), Np(V), and Np(VI) in Aqueous Solution. In: Inorganic Chemistry. Band 48, Nr. 12, 15. Juni 2009, S. 5350–5360, doi:10.1021/ic9003005.
- ↑ Thomas Vercouter, Pierre Vitorge, Badia Amekraz, Christophe Moulin: Stoichiometries and Thermodynamic Stabilities for Aqueous Sulfate Complexes of U(VI). In: Inorganic Chemistry. Band 47, Nr. 6, 1. März 2008, S. 2180–2189, doi:10.1021/ic701379q.
- ↑ I. Ravikumar, Pradyut Ghosh: Recognition and separation of sulfate anions. In: Chemical Society Reviews. Band 41, Nr. 8, 2012, S. 3077, doi:10.1039/c2cs15293b.
- ↑ Radu Custelcean, Jerome Bosano, Peter V. Bonnesen, Vilmos Kertesz, Benjamin P. Hay: Computer‐Aided Design of a Sulfate‐Encapsulating Receptor. In: Angewandte Chemie International Edition. Band 48, Nr. 22, 18. Mai 2009, S. 4025–4029, doi:10.1002/anie.200900108.
- ↑ Chuandong Jia, Biao Wu, Shaoguang Li, Xiaojuan Huang, Qilong Zhao, Qian‐Shu Li, Xiao‐Juan Yang: Highly Efficient Extraction of Sulfate Ions with a Tripodal Hexaurea Receptor. In: Angewandte Chemie International Edition. Band 50, Nr. 2, 10. Januar 2011, S. 486–490, doi:10.1002/anie.201004461.
- ↑ Barbara Elvers, Stephen Hawkins, Wiliam E. Russey: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 25. VCH, Weinheim 1994, ISBN 3-527-20125-4, S. 322 (Eintrag zu 'Strontium and Strontium Compounds').
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 15. VCH, Weinheim/New York 1990, ISBN 3-527-20115-7, S. 195–196 (Eintrag zu 'Lead').
- ↑ Mohammad Hasan Sadeghi, Mohsen Nasr Esfahany: Development of a Safe and Environmentally Friendly Sulfate Process for the Production of Titanium Oxide. In: Industrial & Engineering Chemistry Research. Band 61, Nr. 4, 2. Februar 2022, S. 1786–1796, doi:10.1021/acs.iecr.1c03364.
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 22. VCH, Weinheim 1993, ISBN 3-527-20122-X, S. 618 (Eintrag zu 'Rare Earth Elements').
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 3. VCH, Weinheim 1985, ISBN 3-527-20103-3, S. 330–332 (Eintrag zu 'Barium and Barium Compounds').
- ↑ K.A. Fattah, A. Lashin: Investigation of mud density and weighting materials effect on drilling fluid filter cake properties and formation damage. In: Journal of African Earth Sciences. Band 117, Mai 2016, S. 345–357, doi:10.1016/j.jafrearsci.2016.02.003.
- ↑ Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 4. VCH, Weinheim 1985, ISBN 3-527-20104-1, S. 573–579 (Eintrag zu 'Calcium Sulfate').
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 4. VCH, Weinheim 1985, ISBN 3-527-20104-1, S. 555 (Eintrag zu 'Calcium Sulfate').
- ↑ Yoshiyuki Kojima, Tamotsu Yasue: Synthesis of large plate-like gypsum dihydrate from waste gypsum board. In: Journal of the European Ceramic Society. Band 26, Nr. 4–5, Januar 2006, S. 777–783, doi:10.1016/j.jeurceramsoc.2005.06.018.
- ↑ a b Funda İnceoğlu, Nevin Karamahmut Mermer, Volkan Kırmızı, Gülden Tombaş: Influence of cement with different calcium sulfate phases on cementitious tile adhesive mortars: Microstructure and performance aspects. In: International Journal of Adhesion and Adhesives. Band 104, Januar 2021, S. 102744, doi:10.1016/j.ijadhadh.2020.102744.
- ↑ İlker Bekir Topçu: Properties of heavyweight concrete produced with barite. In: Cement and Concrete Research. Band 33, Nr. 6, Juni 2003, S. 815–822, doi:10.1016/S0008-8846(02)01063-3.
- ↑ Jacquelyn M. Powers, George R. Buchanan, Leah Adix, Song Zhang, Ang Gao, Timothy L. McCavit: Effect of Low-Dose Ferrous Sulfate vs Iron Polysaccharide Complex on Hemoglobin Concentration in Young Children With Nutritional Iron-Deficiency Anemia: A Randomized Clinical Trial. In: JAMA. Band 317, Nr. 22, 13. Juni 2017, S. 2297, doi:10.1001/jama.2017.6846, PMID 28609534, PMC 5815003 (freier Volltext).
- ↑ Hailey I. Kilian, Huijuan Zhang, Mohammad Mahdi Shiraz Bhurwani, Anoop M. Nilam, Daewoon Seong, Mansik Jeon, Ciprian N. Ionita, Jun Xia, Jonathan F. Lovell: Barium sulfate and pigment admixture for photoacoustic and x-ray contrast imaging of the gut. In: Journal of Biomedical Optics. Band 28, Nr. 08, 10. Februar 2023, doi:10.1117/1.JBO.28.8.082803, PMID 36776721, PMC 9917716 (freier Volltext).
- ↑ Jinhua Wen, Darrell Sawmiller, Brendan Wheeldon, Jun Tan: A Review for Lithium: Pharmacokinetics, Drug Design, and Toxicity. In: CNS & Neurological Disorders – Drug Targets. Band 18, Nr. 10, 17. Januar 2020, S. 769–778, doi:10.2174/1871527318666191114095249.
- ↑ Ramadhan Oruch, Mahmoud A. Elderbi, Hassan A. Khattab, Ian F. Pryme, Anders Lund: Lithium: A review of pharmacology, clinical uses, and toxicity. In: European Journal of Pharmacology. Band 740, Oktober 2014, S. 464–473, doi:10.1016/j.ejphar.2014.06.042.
- ↑ G. Steffen Paulekuhn, Jennifer B. Dressman, Christoph Saal: Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database. In: Journal of Medicinal Chemistry. Band 50, Nr. 26, 27. Dezember 2007, S. 6665–6672, doi:10.1021/jm701032y.
- ↑ Niels F. Muller, Rudolf P. Dessing: European Drug Index: European Drug Registrations. 4. Auflage. Routledge, 2022, ISBN 978-1-351-44947-2, S. 117.
- ↑ a b George G. Zhanel, Christopher D. Lawson, Heather Adam, Frank Schweizer, Sheryl Zelenitsky, Philippe R. S. Lagacé-Wiens, Andrew Denisuik, Ethan Rubinstein, Alfred S. Gin, Daryl J. Hoban, Joseph P. Lynch, James A. Karlowsky: Ceftazidime-Avibactam: a Novel Cephalosporin/β-lactamase Inhibitor Combination. In: Drugs. Band 73, Nr. 2, Februar 2013, S. 159–177, doi:10.1007/s40265-013-0013-7.
- ↑ Fahd K. Majiduddin, Isabel C. Materon, Timothy G. Palzkill: Molecular analysis of beta-lactamase structure and function. In: International Journal of Medical Microbiology. Band 292, Nr. 2, 2002, S. 127–137, doi:10.1078/1438-4221-00198.
- ↑ a b Susan J. Keam: Sulbactam/Durlobactam: First Approval. In: Drugs. Band 83, Nr. 13, September 2023, S. 1245–1252, doi:10.1007/s40265-023-01920-6.
- ↑ Michael J Satlin: Languid Uptake of Ceftazidime-Avibactam for Carbapenem-Resistant Gram-Negative Infections and Continued Reliance on Polymyxins. In: Clinical Infectious Diseases. Band 72, Nr. 4, 16. Februar 2021, S. 622–625, doi:10.1093/cid/ciaa065.
- ↑ Bhagu R. Bhavnani, Frank Z. Stanczyk: Pharmacology of conjugated equine estrogens: Efficacy, safety and mechanism of action. In: The Journal of Steroid Biochemistry and Molecular Biology. Band 142, Juli 2014, S. 16–29, doi:10.1016/j.jsbmb.2013.10.011.
- ↑ Suresh P. Sulochana, Muzeeb Syed, Devaraj V. Chandrasekar, Ramesh Mullangi, Nuggehally R. Srinivas: Clinical Drug–Drug Pharmacokinetic Interaction Potential of Sucralfate with Other Drugs: Review and Perspectives. In: European Journal of Drug Metabolism and Pharmacokinetics. Band 41, Nr. 5, Oktober 2016, S. 469–503, doi:10.1007/s13318-016-0335-4.
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 22. VCH, Weinheim 1993, ISBN 3-527-20122-X, S. 653 (Eintrag zu 'Reactive Dyes').
- ↑ a b Guoqiang Cai, Liangxi Sun, Jindan Wu, Jiping Wang: Influence of Nonionic Surfactant on Hydrolysis of Vinyl Sulfone Reactive Dye. In: Journal of Surfactants and Detergents. Band 18, Nr. 6, November 2015, S. 1127–1135, doi:10.1007/s11743-015-1726-2.
- ↑ a b J. Heyna: Reactive Dyes Containing Vinylsulfonyl Groups. In: Angewandte Chemie International Edition in English. Band 2, Nr. 1, Januar 1963, S. 20–23, doi:10.1002/anie.196300201.
- ↑ W. J. O’Brien: A Study of Lithopone. In: The Journal of Physical Chemistry. Band 19, Nr. 2, 1. Februar 1915, S. 113–144, doi:10.1021/j150155a002.
- ↑ Han Gao, Shuxue Yang, Danjun Mao, Mingce Long, Xiaolei Qu: Significant zinc release from widely-used commercial lithopone pigments under solar irradiation. In: Environmental Pollution. Band 292, Januar 2022, S. 118352, doi:10.1016/j.envpol.2021.118352.
- ↑ Anthony D. Covington: Modern tanning chemistry. In: Chemical Society Reviews. Band 26, Nr. 2, 1997, S. 111, doi:10.1039/cs9972600111.
- ↑ Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 8. VCH, Weinheim/New York 1987, ISBN 3-527-20108-4, S. 345–346 (Eintrag zu 'Detergents').
- ↑ Valerie C. Robinson, Wilma F. Bergfeld, Donald V. Belsito, Ronald A. Hill, Curtis D. Klaassen, James G. Marks, Ronald C. Shank, Thomas J. Slaga, Paul W. Snyder, F. Alan Andersen: Final Report of the Amended Safety Assessment of Sodium Laureth Sulfate and Related Salts of Sulfated Ethoxylated Alcohols. In: International Journal of Toxicology. Band 29, 4_suppl, Mai 2010, S. 151S–161S, doi:10.1177/1091581810373151.
- ↑ José G. Ortiz-Tena, Doris Schieder, Volker Sieber: Carrageenan and More: Biorefinery Approaches with Special Reference to the Processing of Kappaphycus. In: Tropical Seaweed Farming Trends, Problems and Opportunities. Springer International Publishing, Cham 2017, ISBN 978-3-319-63497-5, S. 155–164, doi:10.1007/978-3-319-63498-2_10.
- ↑ Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 2. VCH, Weinheim 1985, ISBN 3-527-20102-5, S. 252–255 (Eintrag zu 'Ammonium Compounds').
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 15. VCH, Weinheim/New York 1990, ISBN 3-527-20115-7, S. 623 (Eintrag zu 'Magnesium Compounds').
- ↑ a b Fritz Ullmann: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 7. VCH, Weinheim/New York 1986, ISBN 3-527-20107-6, S. 578–579 (Eintrag zu 'Copper Compounds').
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann’s encyclopedia of industrial chemistry. 5. Auflage. Band 21. VCH, Weinheim/New York 1991, ISBN 3-527-20121-1, S. 108 (Eintrag zu 'Platinum Group Metals and Compounds').
- ↑ Frank J Schenck, Patrick Callery, Peter M Gannett, Jonathan R Daft, Steven J Lehotay: Comparison of Magnesium Sulfate and Sodium Sulfate for Removal of Water from Pesticide Extracts of Foods. In: Journal of AOAC INTERNATIONAL. Band 85, Nr. 5, 1. September 2002, S. 1177–1180, doi:10.1093/jaoac/85.5.1177.
- ↑ R. T. Sheen, H. L. Kahler, E. M. Ross, W. H. Betz, L. D. Betz: Turbidimetric Determination of Sulfate in Water. In: Industrial & Engineering Chemistry Analytical Edition. Band 7, Nr. 4, 1. Juli 1935, S. 262–265, doi:10.1021/ac50096a022.
- ↑ Eberhard Schweda, Gerhart Jander, Ewald Blasius: Jander/Blasius anorganische Chemie. 1: Theoretische Grundlagen und Qualitative Analyse. 19., völlig neu bearbeitete Auflage. Hirzel Verlag, Stuttgart 2022, ISBN 978-3-7776-3009-0, S. 249–250.
Anmerkungen
[Bearbeiten | Quelltext bearbeiten]- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Dihexylsulfat: CAS-Nr.: 7722-57-8, PubChem: 14747182, Wikidata: Q125176908.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Pregnenolonsulfat: CAS-Nr.: 1247-64-9, PubChem: 105074, ChemSpider: 94802, Wikidata: Q7239912.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu N-Acetyllactosamin: CAS-Nr.: 32181-59-2, EG-Nr.: 636-520-5, ECHA-InfoCard: 100.164.310, PubChem: 439271, ChemSpider: 7975931, Wikidata: Q19903180.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Octylsulfat: CAS-Nr.: nicht vergeben, PubChem: 5121662, ChemSpider: 4296365, Wikidata: Q27156569.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Decylsulfat: CAS-Nr.: nicht vergeben, PubChem: 5003059, ChemSpider: 4182760, Wikidata: Q27156567.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Isooctylsulfat: CAS-Nr.: 309941-87-5, Wikidata: Q125176927.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Cholesterolsulfat: CAS-Nr.: 1256-86-6, PubChem: 65076, ChemSpider: 58586, DrugBank: DBDB01990, Wikidata: Q27075985.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Zetekitoxin: CAS-Nr.: 9061-57-8, PubChem: 76853098, Wikidata: Q82892962.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Homomethionin: CAS-Nr.: 25148-30-5, PubChem: 10329619, ChemSpider: 8505080, Wikidata: Q27122197.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Homophenylalanin: CAS-Nr.: 943-73-7, EG-Nr.: 213-403-3, ECHA-InfoCard: 100.012.185, PubChem: 2724505, ChemSpider: 2006639, Wikidata: Q27120508.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Isoscutellarin: CAS-Nr.: 62023-92-1, PubChem: 11996857, Wikidata: Q125176930.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Hypolaetin: CAS-Nr.: 27696-41-9, PubChem: 5281648, ChemSpider: 4444967, Wikidata: Q15411029.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Lactucin: CAS-Nr.: 1891-29-8, PubChem: 442266, ChemSpider: 717609, Wikidata: Q6469057.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu 1-O-Coumaroylglucose: CAS-Nr.: 7139-64-2, PubChem: 14158117, ChemSpider: 10394547, Wikidata: Q27139663.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu 1-O-Caffeoylglucose: CAS-Nr.: 14364-08-0, PubChem: 6124135, ChemSpider: 4445075, Wikidata: Q27105318.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Cylindrospermopsin: CAS-Nr.: 143545-90-8, EG-Nr.: 802-929-4, ECHA-InfoCard: 100.229.780, PubChem: 135565888, ChemSpider: 26396261, Wikidata: Q3008693.
- ↑ a b c Externe Identifikatoren von bzw. Datenbank-Links zu Monomethylsulfat: CAS-Nr.: 21228-90-0, PubChem: 4694097, ChemSpider: 3881721, Wikidata: Q27125654.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Ethylchlorsulfit: CAS-Nr.: 6378-11-6, PubChem: 80784, Wikidata: Q82862976.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Phosphoadenosinphosphat: CAS-Nr.: 1053-73-2, PubChem: 159296, ChemSpider: 140102, DrugBank: DBDB01812, Wikidata: Q2825485.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu para-Nitrophenylsulfat: CAS-Nr.: 1080-04-2, PubChem: 80349, ChemSpider: 72581, Wikidata: Q27116472.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu N-Hydroxysuccinimidsulfat: CAS-Nr.: 127007-81-2, PubChem: 23447253, Wikidata: Q82542259.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Dicyclohexylethylamin: CAS-Nr.: 7175-49-7, EG-Nr.: 230-534-1, ECHA-InfoCard: 100.027.759, PubChem: 23563, Wikidata: Q72474281.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Di-tert-butylmalonat: CAS-Nr.: 541-16-2, EG-Nr.: 208-769-6, ECHA-InfoCard: 100.007.973, PubChem: 68324, ChemSpider: 61619, Wikidata: Q27268154.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Diphenylsulfat: CAS-Nr.: 4074-56-0, PubChem: 14228015, ChemSpider: 10738548, Wikidata: Q82439695.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Manganlinolat: CAS-Nr.: 646-00-4, EG-Nr.: 211-459-3, ECHA-InfoCard: 100.010.419, PubChem: 20839547, ChemSpider: 20144576, Wikidata: Q17320557.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Durlobactam: CAS-Nr.: 1467829-71-5, PubChem: 89851852, DrugBank: DBDB16704, Wikidata: Q100151634.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Ammoniumlaurylethersulfat: CAS-Nr.: 32612-48-9, EG-Nr.: 608-760-0, ECHA-InfoCard: 100.125.985, PubChem: 61913, Wikidata: Q21547002.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Magnesiumlaurylethersulfat: CAS-Nr.: 62755-21-9, EG-Nr.: 613-078-1, ECHA-InfoCard: 100.117.245, PubChem: 44150971, Wikidata: Q6731396.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Natriumrhodizonat: CAS-Nr.: 523-21-7, EG-Nr.: 208-340-3, ECHA-InfoCard: 100.007.584, GESTIS: 104671, PubChem: 68225, ChemSpider: 61528, Wikidata: Q3939892.
- ↑ Externe Identifikatoren von bzw. Datenbank-Links zu Bariumrhodizonat: CAS-Nr.: 16833-52-6, PubChem: 85605, Wikidata: Q81992699.